This is revision of a previous post.
Building Processes:
This post is mainly on building processes for a non-turnkey system such as the Medinfo Hematos IIG software that I have worked with in several countries, but there will be a few words about turnkey systems for general laboratories.
This has been a collaborative effort between the software vendor’s engineers, my Super Users, and myself. This pluralistic approach has been most productive.
A turnkey system has pretty much already defined most of the basic processes—those have been specifically approved by a regulatory agency such as US FDA. There is little customization except formatting screen and reports. Instrument interfaces are also mainly predefined. This requires much less thought and planning than a custom-built system designed on the sites actual workflows, but it can be an exercise of putting a round peg in a square hole. You don’t always get what you want.
In the locations where I collaborated in setting up the Medinfo Hematos IIG program, we did not follow US FDA but mainly the Council of Europe CE standards since this was much more customizable. Additionally, we could modify and add additional criteria specific to our country and region (e.g. rules for donor qualification for local pathogens). This has always been my preferred approach.
Start with a frame of reference (CE) and then try to optimize it for our local needs.
I recommend the Council of Europe CE since it is more flexible and extensible. Those subject to the US FDA has many fewer approved options in the preparation of blood components (e.g. prohibiting the use of pooled buffy coat platelets, automated blood component production such as Reveos, and use of world-class pathogen-inactivation technologies such as Mirasol.)
If you invested the time to make a detailed workflow across all current processes and tests, much of this can be readily translated into the software processes, but first you must study the flows and determine where you can optimize them. This requires that you study the options in the new software to see what you can use best.
I always liked Occam’s Razor, i.e. “ntia non sunt multiplicanda praeter necessitatem,”—the simpler the better as long as it meets your needs. If the manual processes are working well and can be translated into the new system, do so. If they need changes for optimization, then do so only if necessary.
Most of my career has been spent overseas with staff from many different countries and backgrounds, most of whom were not native in English. The wording of the processes is very important. Think of the additional obstacle of working with a complicated software in your non-mother tongue! Also consider the differences between American English, British English, and international English. I always made the Super Users read my proposed specifications and then asked them to repeat what I wrote/said.
There were many surprises discovered. I think of the Aesop’s fable about the mother who gave birth to an ugly baby looking like a monkey. Still, to the mother her baby was the most beautiful baby and she entered him into a beauty contest. In other words, to the mother her child is perfect!
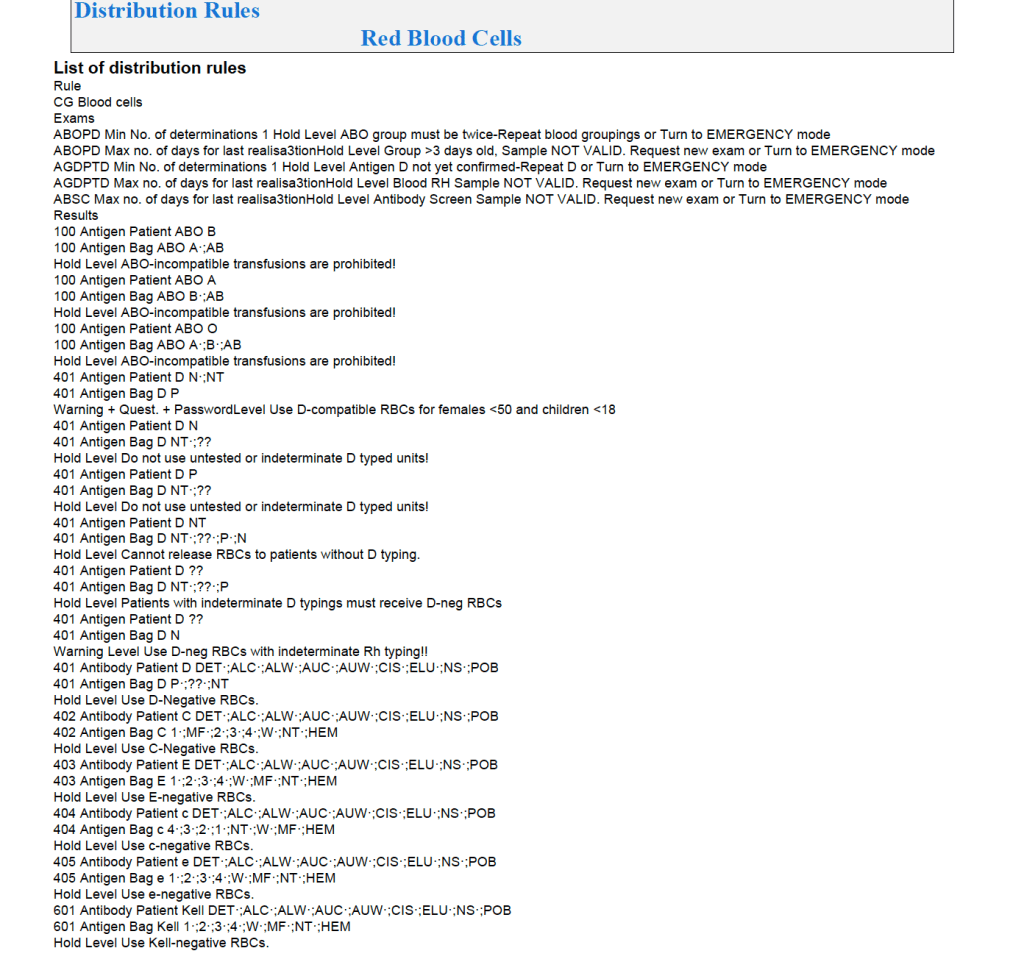
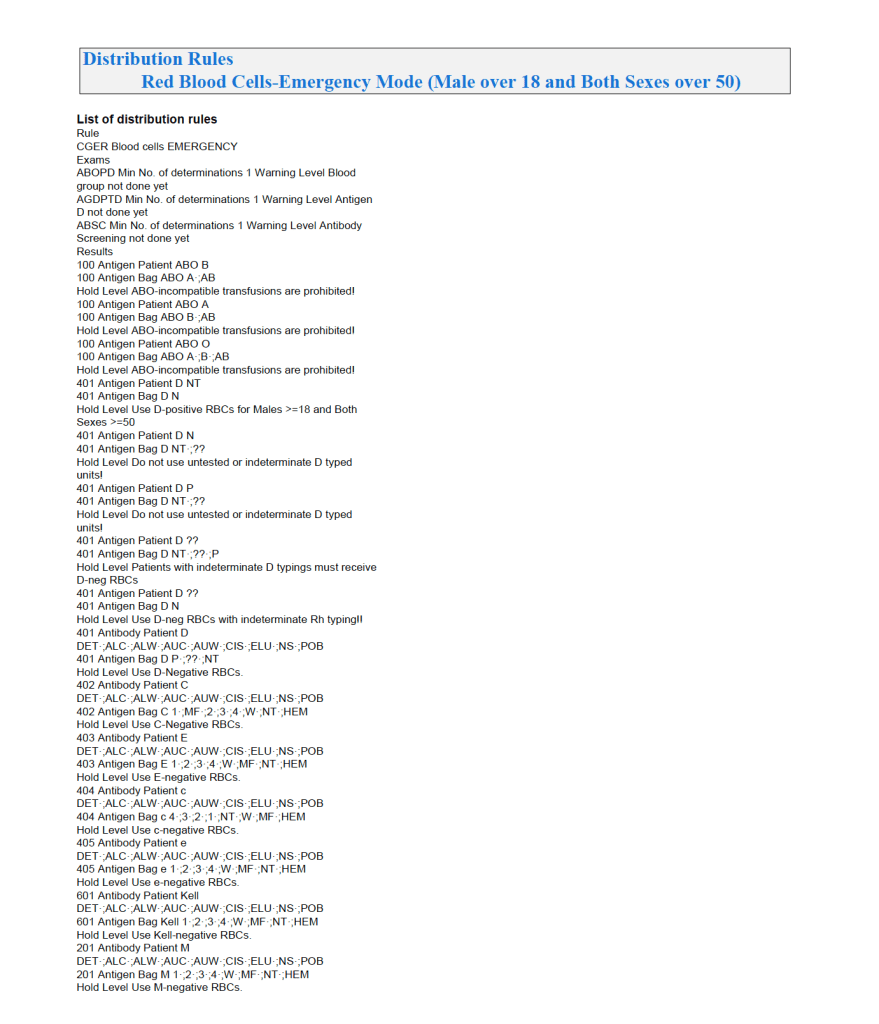
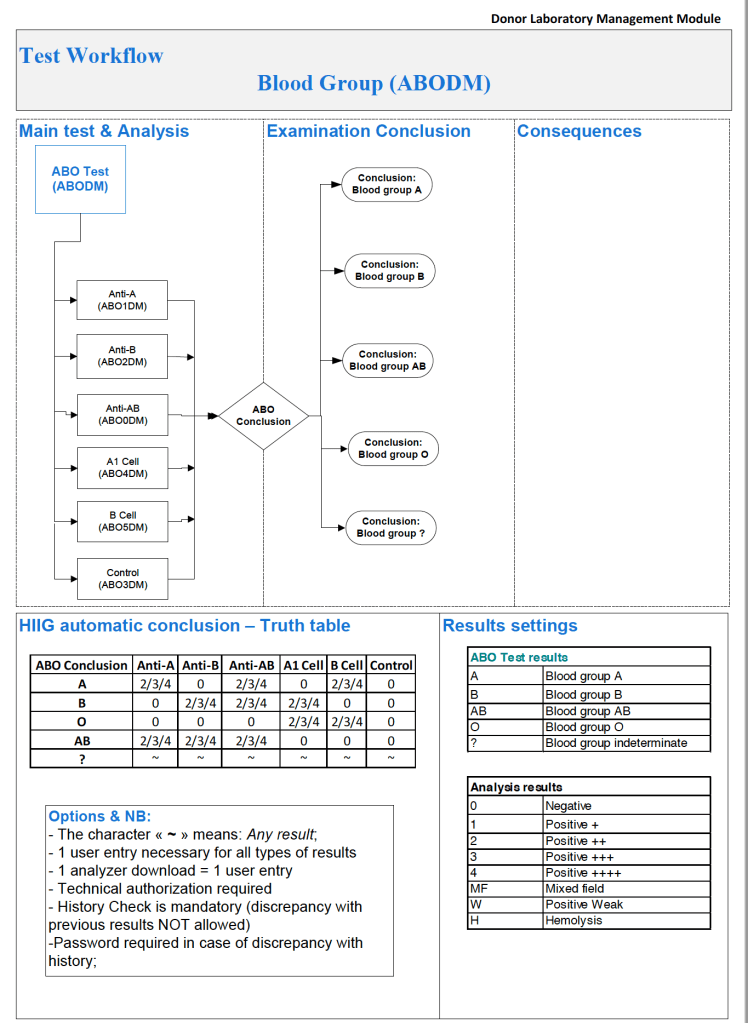
It is most important to use the manufacturer’s recommendations to build tests and for the special automated processing and pathogen-inactivation processes. For example, we had multiple ABO and D typing tests—they did not necessarily agree on what were acceptable results for automated release of results. The same is true for many other tests.
Example: One method for Rh(D) typing stated that only results in {0, 2+, 3+, 4+} were acceptable—all other results required manual review and/or additional testing. Another only accepted results in {0,3,4}. Thus we had to build separate D typing processes for each methodology.
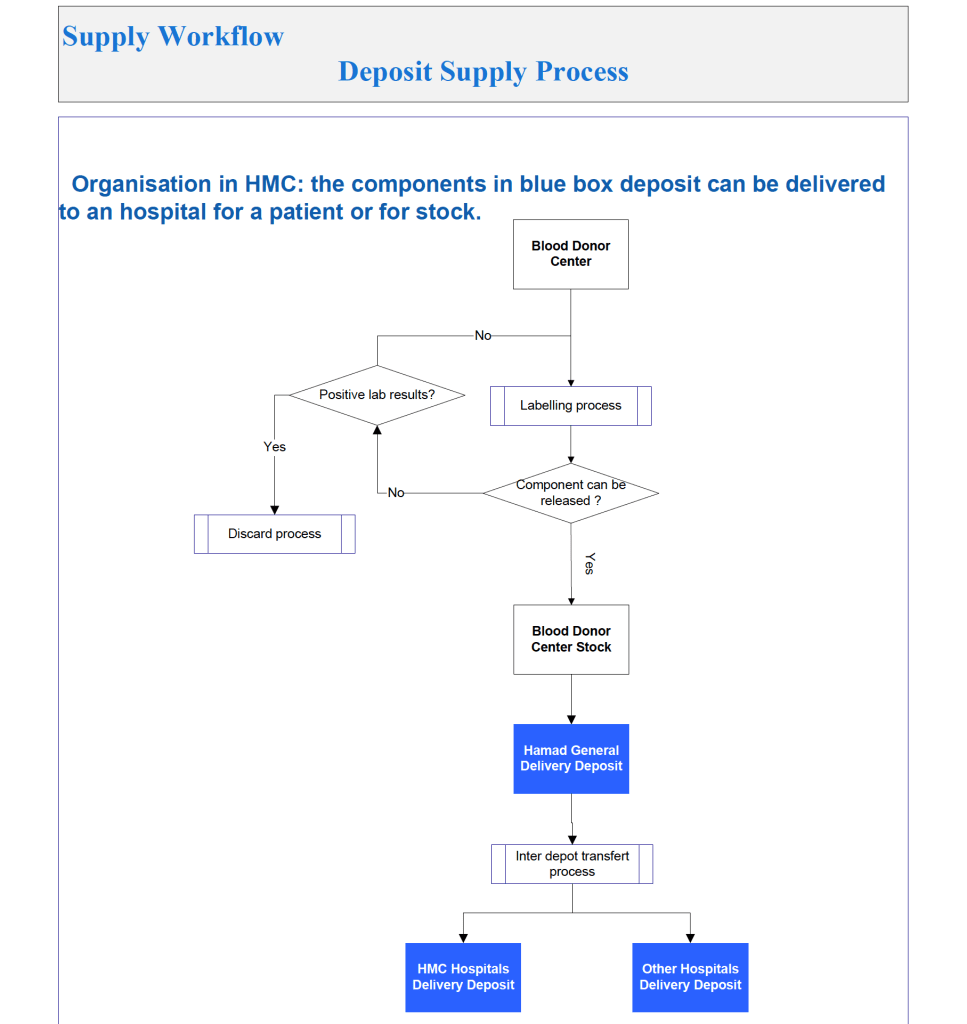
Another consideration is whether to offer all the processes globally or restricted to one site. I favor allowing access to all methodologies at all sites—in case of a disaster where tests had to performed at another site. This means that if you send an order over an interface from the hospital system to the blood bank system, that at the receiving (blood bank) end, you can choose which methodology to use, i.e. it is not a one-to-one mapping but rather a one to many mapping.
If we changed equipment at one site to that used at another site, we didn’t have to modify our software to accommodate this. Even if you didn’t have the equipment or reagents at one site, you could always build it into the system and not activate the settings until needed.
Finally, the issue of middleware. Many instruments offer this, but one faces the problem about support and regression errors when you either update the middleware software or the blood bank computer software. Medinfo itself can serve as the middleware so there is less chance of errors when updating the software. In fact, I never have used any middleware when using Medinfo.
Instrument interfaces will be a future topic.