Anyone reviewing antibody panels, especially in the Middle East/Gulf region, encounters many panels for which no antibody specificity is identified. As a transfusion medicine physician who often got called during the night for release of RBCs for patients with “nonspecific” pattern, this was a big headache.
Is it “nonspecific” because there isn’t a clinically significant antibody OR the technologist did not perform the testing or its interpretation correctly? Does it need further testing? Do I release blood components at this time?
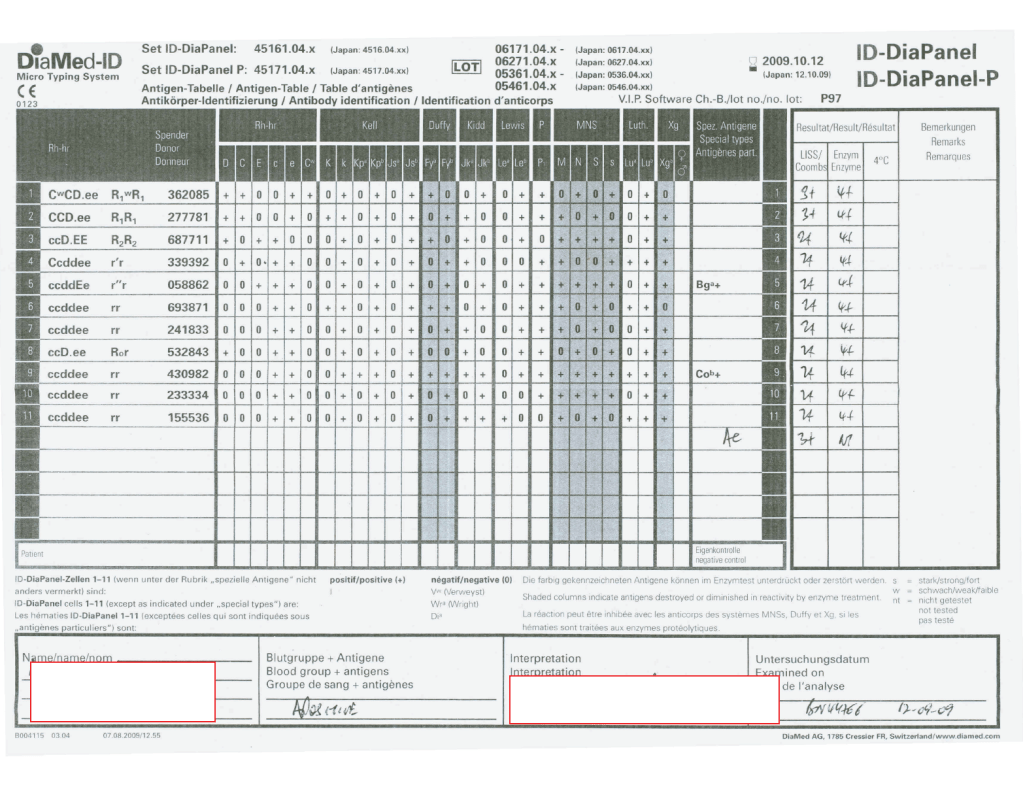
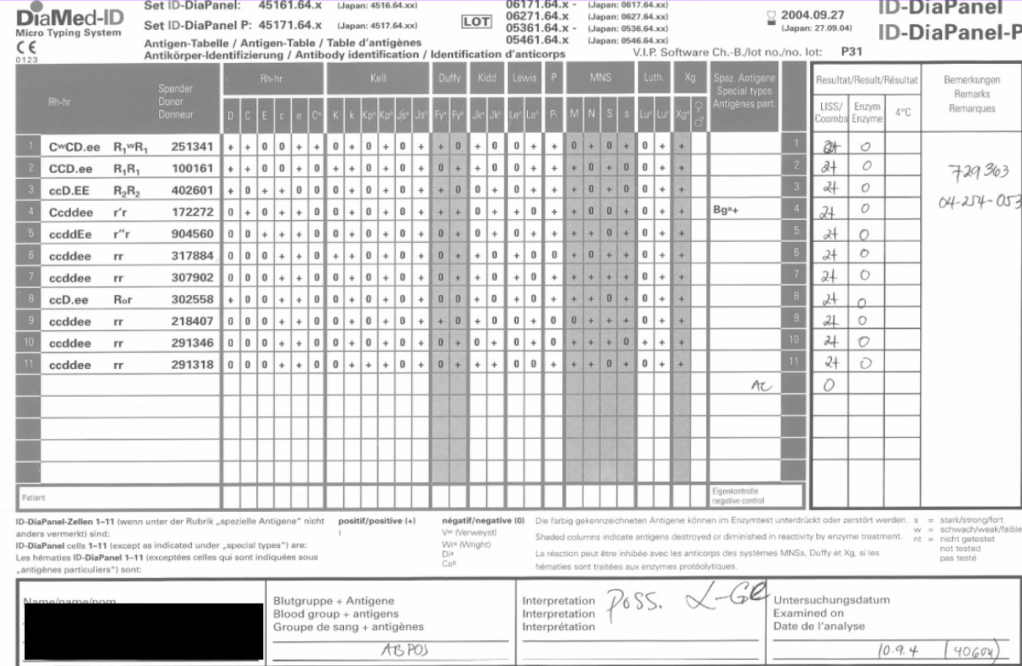
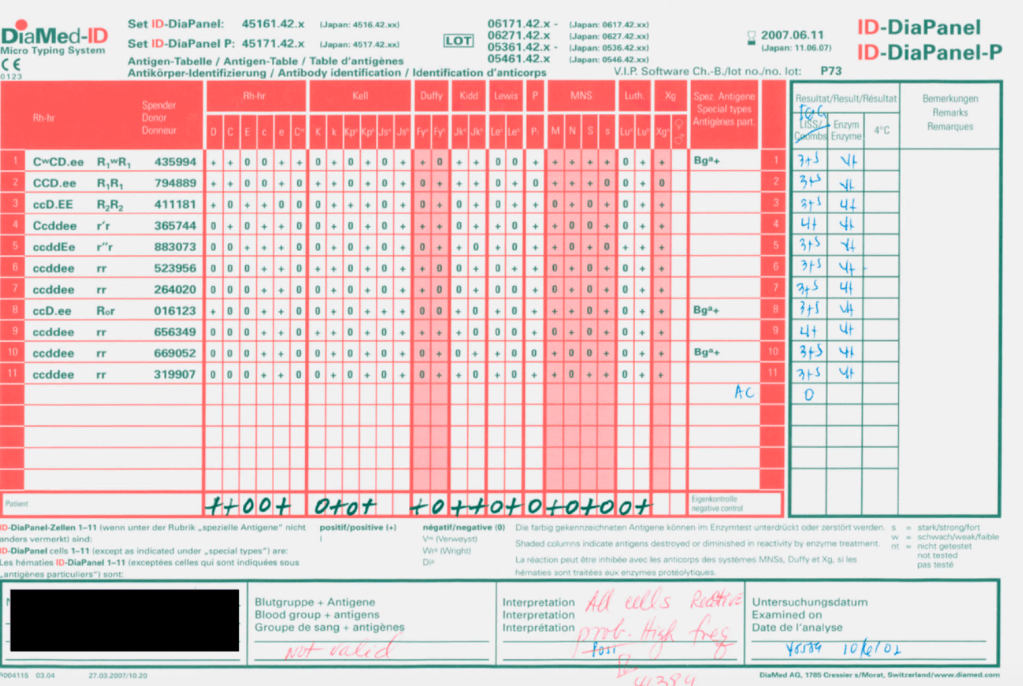
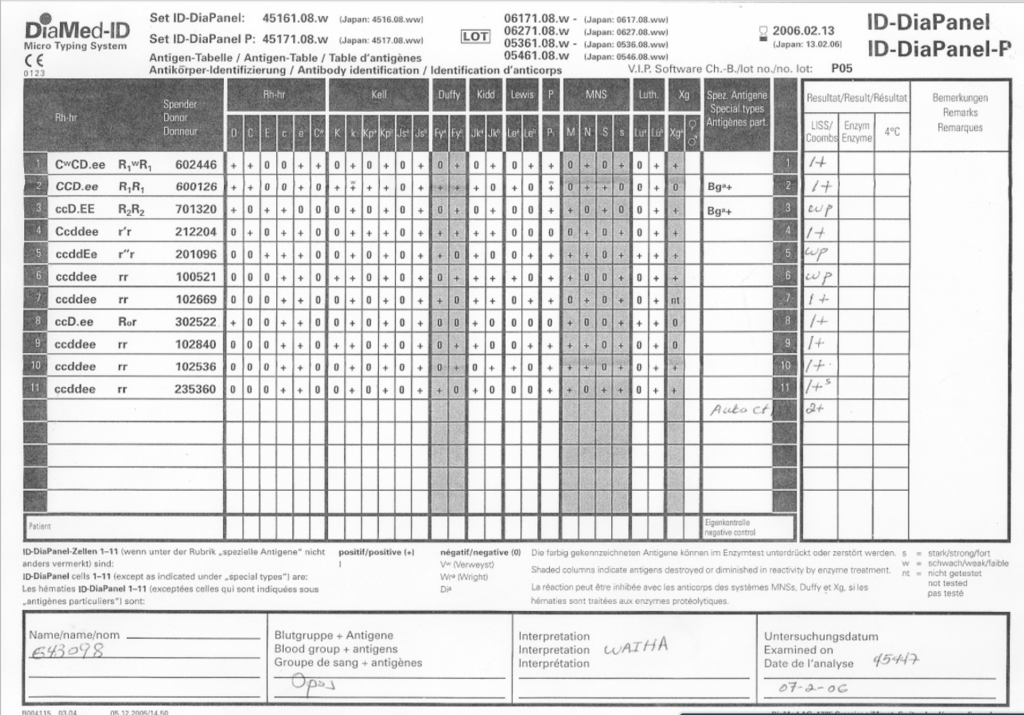
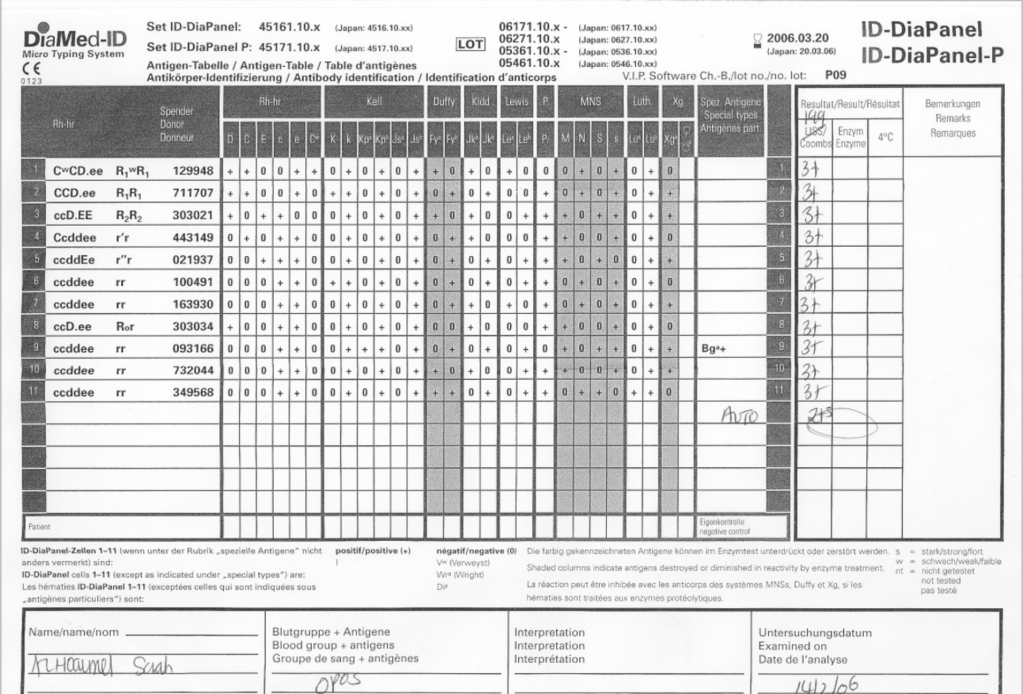
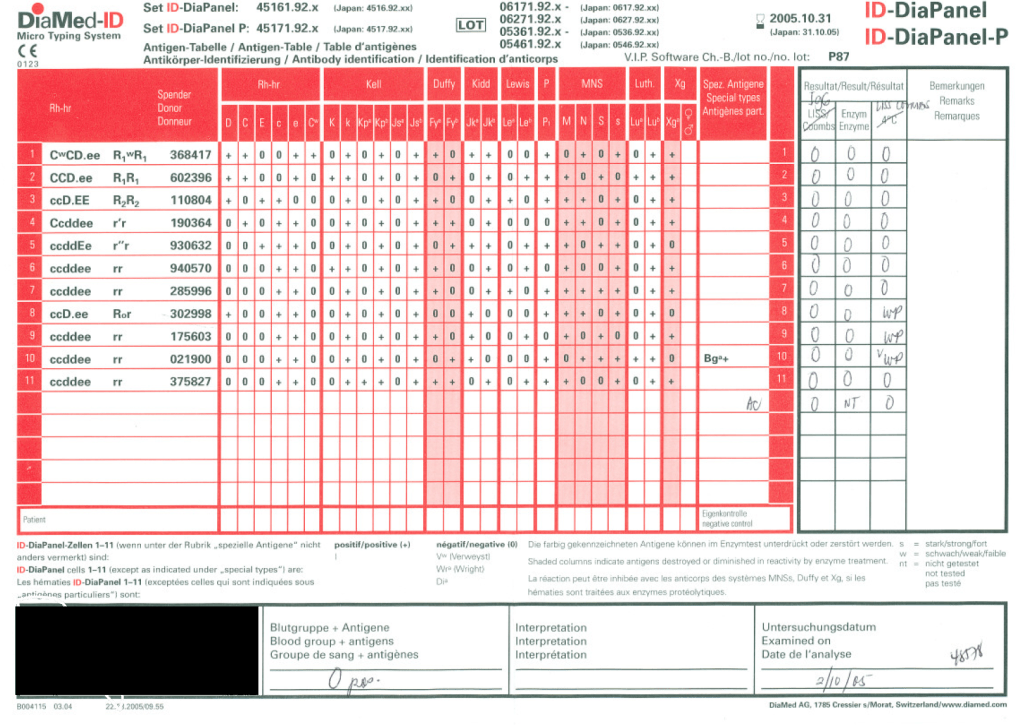
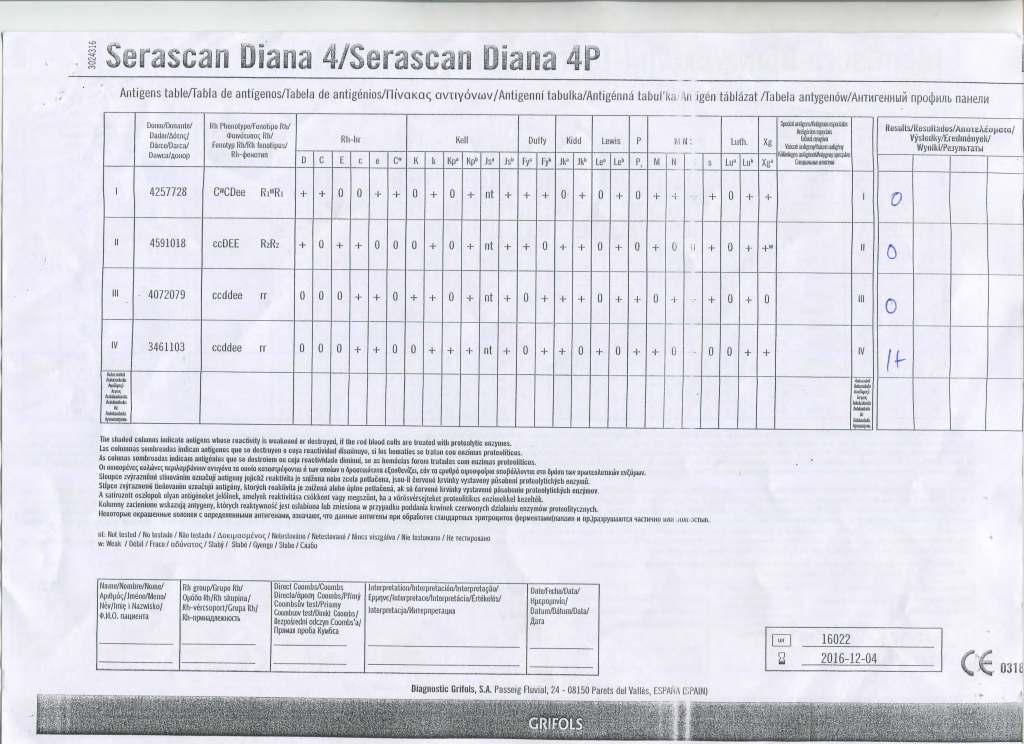
I have attached an example of an antibody panel that has a few weak to 1+ reactions using a gamma-whole-molecule IgG AHG reagent. Notice that they are enzyme-labile.

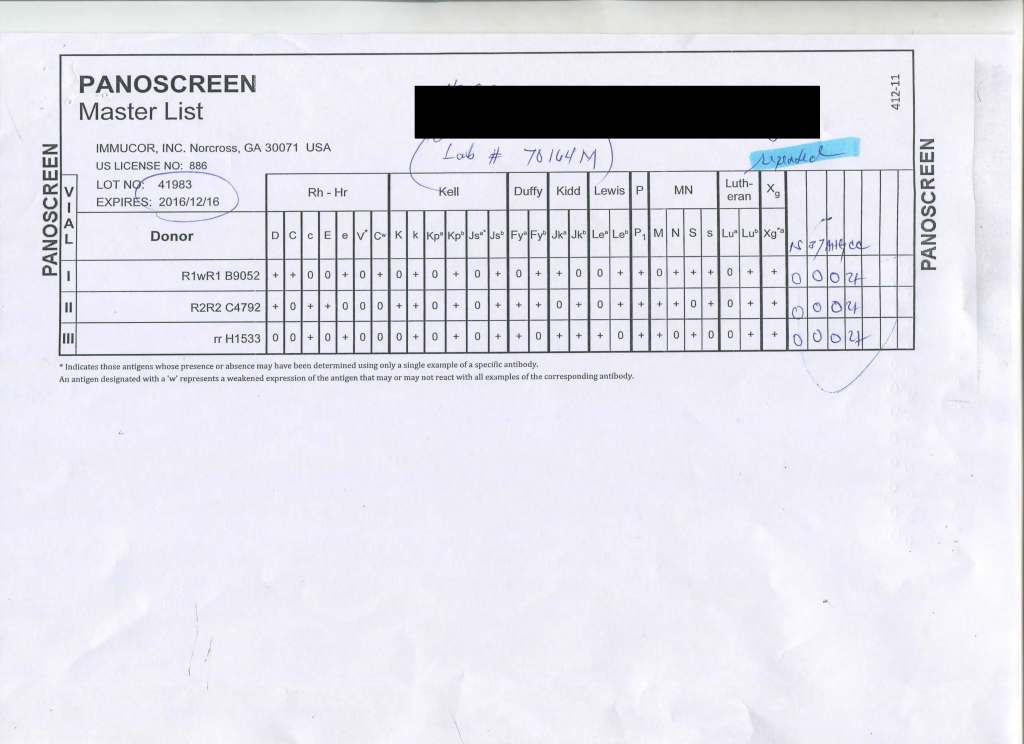
I have worked in this region both before and after gel/glass bead technology was introduced. In the good old days, I formerly only used a gamma heavy-chain specific AHG in tubes with LISS. Nonspecific reactions were few.
When we adopted gel and at some sites glass bead columns, there was only a choice between polyspecific AHG and whole-molecule IgG AHG. The rate of non-specificity soared from less than 10% to over 30% of panels. The reactions were reproducible over multiple technologists at multiple sites.
When I repeated the same specimen against a manual tube panel using gamma-heavy-chain specific AHG, the reactions disappeared in the majority of the cases. Why?
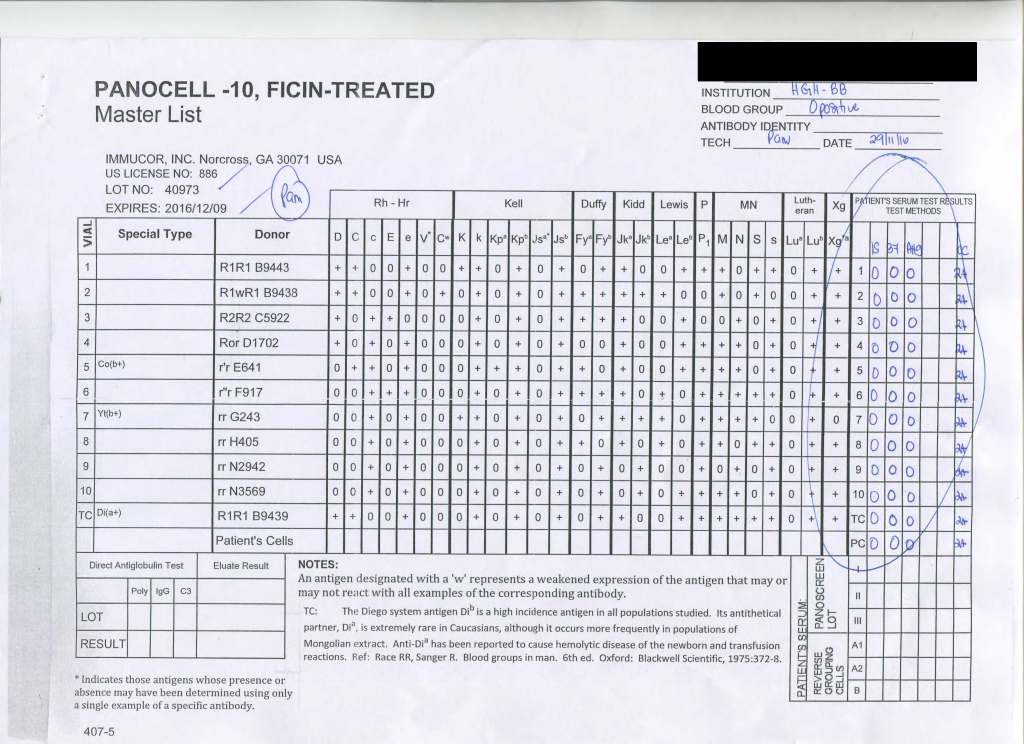
Heavy chain specific reagents do not detect light chains. Light chains are the same in IgM and IgG antibodies. Both polyspecific and whole molecule IgG AHG detect light chains. Thus, high-thermal-amplitude cold antibodies may be detected with the latter but are unlikely to react with heavy chain specific reagents.
When I used manual tube reagents, I would have missed these reactions, but I do not recall this having clinical consequences except in very rare circumstances (refer to my previous post about the anti-Jka only detectable with polyspecific reagents—I have seen 3 in 30 years).
I have asked various gel/glass bead manufacturers to provide me with heavy-chain gamma-specific AHG, but none have agreed.
Normally, for automation, I use IgG-whole molecule AHG. If the reactions are nonspecific, we repeat the testing manually using the gamma-heavy-chain-specific AHG. If negative, I ignore the nonspecific reactions and use the same gamma-heavy-chain-specific AHG for full antiglobulin-phase crossmatching.
In general, with nonspecific reactions, I recommend the following:
- Always do enzyme panels, sometimes with both papain and ficin reagents: many Rh antibodies are optimally detected only at enzyme (example: R1R1 with apparent anti-E at AHG but the anti-c only seen at enzyme)
- Perform an extended Rh/Kell/Duffy/Kidd/Kell/MNSs/P1 phenotype and specifically check for those negative typing results AND for dosage (could the antibody only be detected in homozygous cells like many anti-M are?).
- Perform classical room temperature, 37C, and finally AHG phase testing. Routinely I do not do this since antibodies not detected at 37C are unlikely to be clinically significant. Sometimes, the AHG phase reactivity is a cold antibody of high-thermal amplitude.
- If the Jka or Jkb antigen typings are negative, repeat using a polyspecific AHG reagent.
- Use additional panels from multiple manufacturers. Some reagents detect more nonspecific reactions than others.
- Try other potentiators than LISS such as PEG.
- Check the outdate of the panel and reagents: if less than 1 week remaining, consider repeating with fresh reagents and getting a new patient sample.
Finally, if you still cannot define the specificity, consider repeating after several days. Maybe it is a newly emerging or an anamnestic response.
I emphasize as a physician, I do not care to see all possible antibodies present in the specimen but rather only those likely to be clinically significant.