Skip to content
Open Menu
Blog (Home)
Transfusion Medicine
Blood Bank IT
COVID-19 Plasma CCP
Plasma Fractionation
About me
Work with me
Contact me
Search
Search for:
Close
Dr. Zeyd Merenkov
Transfusion Medicine, Blood Bank IT, Pathogen Inactivation, Plasma Fractionation, COVID-19 Convalescent Plasma Production
Tag:
Donor Processing
Automated and manual component processing including Reveos, Mirasol, PAS
Traceability
Image
18th Jun 2026
14th Dec 2025
drzeyd
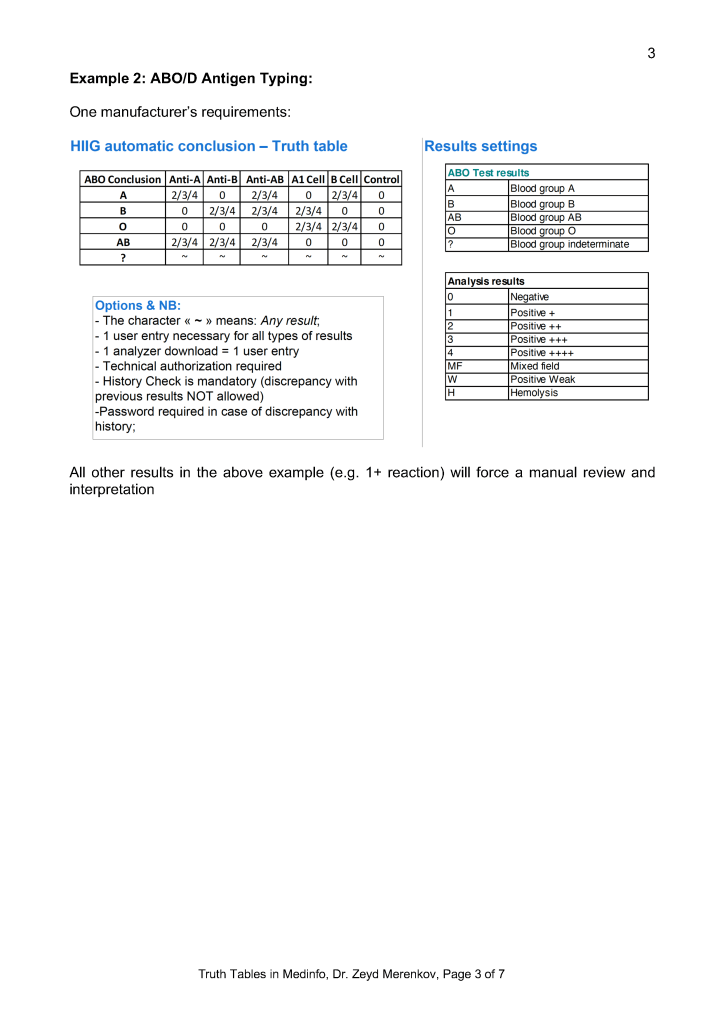
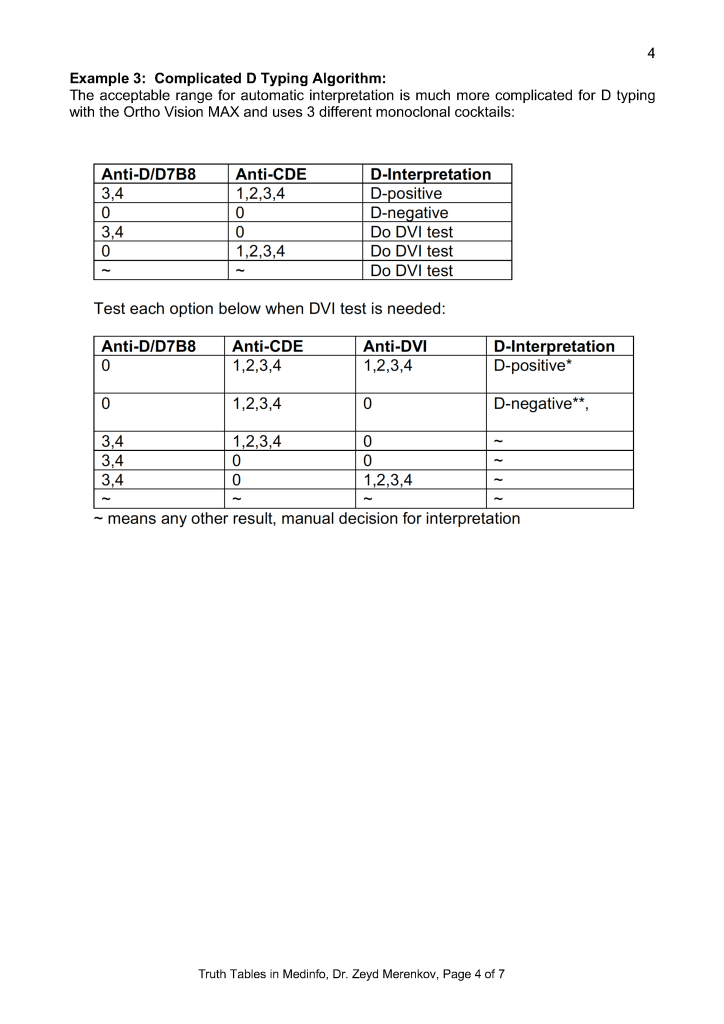
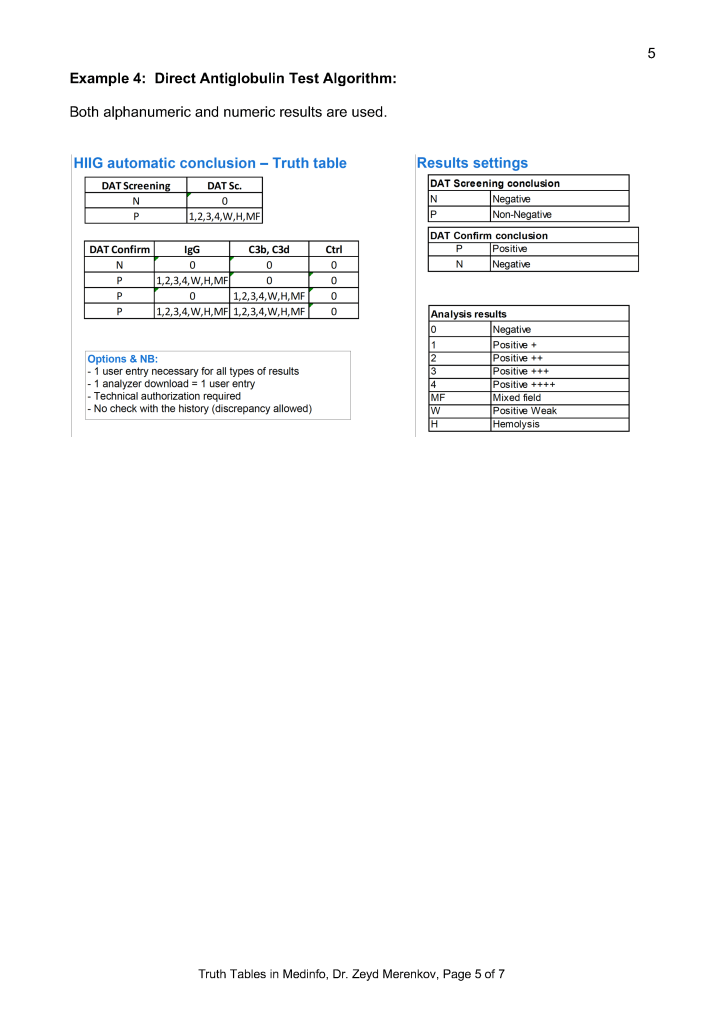
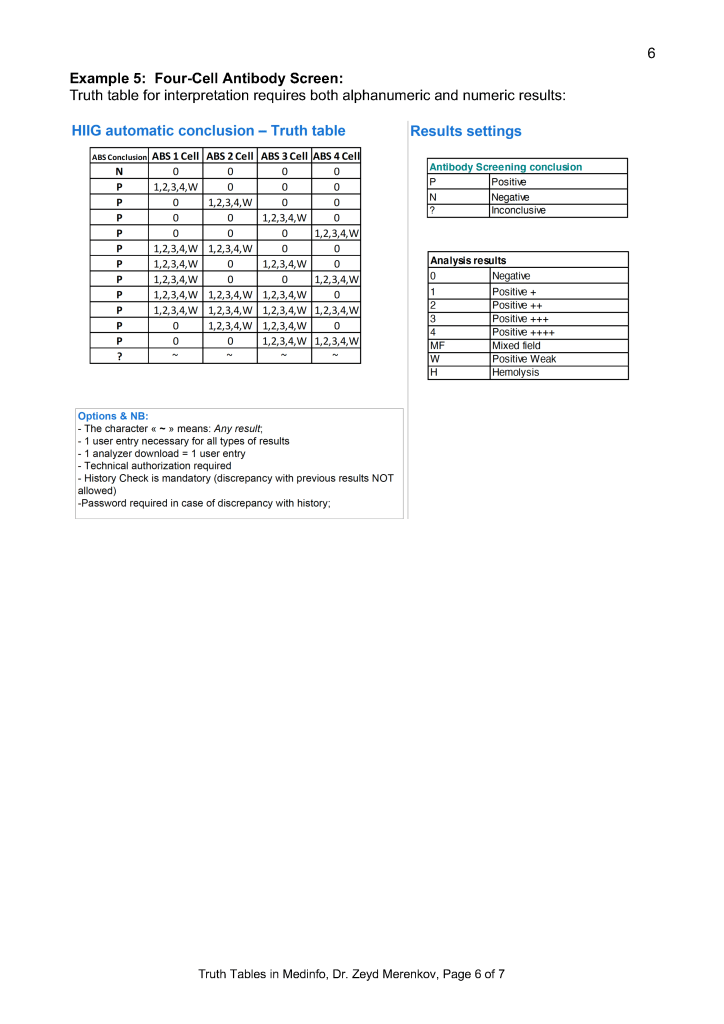
Middleware and Truth Tables
17th Jun 2026
12th Dec 2025
drzeyd
Variances in Transfusion Medicine
Image
10th Jun 2026
6th Dec 2025
drzeyd
Teaching Transfusion Medicine
31st May 2026
24th Nov 2025
drzeyd
Terms of Reference for a Blood Center
30th May 2026
25th Nov 2025
drzeyd
Storage of Frozen Plasma
Image
29th May 2026
22nd Nov 2025
drzeyd
Selecting Platelets
Image
28th May 2026
21st Nov 2025
drzeyd
Donor Screening for New Pathogen
Image
25th May 2026
drzeyd
Writing Transfusion Documentation
22nd May 2026
14th Nov 2025
drzeyd
Massive Transfusion Protocol Wishlist
Image
20th May 2026
12th Nov 2025
drzeyd
Posts navigation
Older Posts
Back to top
Subscribe
Subscribed
Dr. Zeyd Merenkov
Join 41 other subscribers.
Sign me up
Already have a WordPress.com account?
Log in now.
Dr. Zeyd Merenkov
Subscribe
Subscribed
Sign up
Log in
Report this content
View site in Reader
Manage subscriptions
Collapse this bar
Loading Comments...
Write a Comment...
Email (Required)
Name (Required)
Website