


Any or all test methodologies




Principle:
Syphilis, caused by the spirochete Treponema pallidum (T. pallidum), is most often acquired after sexual contact with an infected individual. Syphilis can also be transmitted from mother to child or, rarely, transmitted by transfusion of blood or blood components from donors with active syphilis.
There are two different types of serologic assays for syphilis: nontreponemal assays and treponemal assays:
Nontreponemal assays (e.g. VDRL, RPR, ART) are nonspecific and detect “reagin” antibodies directed against an antigen called cardiolipin that is present in a variety of tissues. Antibodies to cardiolipin appear in the serum of persons with active syphilis or with other medical conditions. However, some individuals who were previously infected with syphilis but successfully treated maintain low levels of antibody to cardiolipin for a long time.
Treponemal assays include enzyme immunoassays (EIA), fluorescent treponemal antibody “absorbed” assays (FTA-ABS), Treponema pallidum microhemagglutination assays (MHA-TPA) and Treponema pallidum particle agglutination assays (TP-PA). Treponemal assays test for antibodies to antigens that are specific to treponemes. Treponemal assays are most useful in identifying recent and past syphilis infections. They are not generally useful in monitoring the response to antibiotic therapy. With some exceptions, positive results of tests for specific treponemal antibodies remain positive throughout an individual’s life regardless of whether the individual is currently infected or has been cured following successful treatment. Retesting sera that are reactive in nontreponemal assays using a specific treponemal test is valuable in distinguishing true-positive results that indicate active syphilis infection from biological false-positive results due to other conditions.
Current testing requirements for syphilis are found in 21 CFR 610.40(a)(2). Individuals who test reactive with a screening test for syphilis must be deferred (21 CFR 610.41(a)) and notified of their deferral (21 CFR 630.40). You must further test each donation found to be reactive by a donor screening test, except you are not required to perform further testing of a donation found to be reactive by a treponemal screening test for syphilis
Policy:
Reference:
Recommendations for Screening, Testing and Management of Blood Donors and Blood and Blood Components Based on Screening Tests for Syphilis—Guidance for Industry, U.S. Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research CBER, December, 2020
This is non-binding CBER guidance is a complicated algorithm that involves using treponemal and non-treponemal assays. Re-entry pathway options are also provided. I will be posting a summary and its implementation into my previous donor marker testing algorithms. Please see attached PDF link.
These are my specifications used in April, 2020 at HMC Qatar for setting up the CCP processes in Medinfo Hematos IIG:


I prepared the following plan for a CCP program for HMC Qatar in March, 2020. The workflow is divided into four (4) modules:
Module 1:
Module 2:
Module 3:
Module 4:
Workflow Considerations:
Logistics:
Information Technology:
It now has been over eight 8 months since I prepared the CCP workflow in Medinfo. It was built on the framework of the manual CCP process including donor prescreening with an abbreviated donor questionnaire. It was really quite simple and used the donor and patient modules to create quarantine areas for donor screening, collection, processing, and hospital patient blood bank release.
Here are my current comments on the process:
Donor Qualification:
I would still exclude malaria and HTLV from the donor questionnaire and would update to UDQ 2.1. Since these donors have recovered from a potentially life-threatening illness, I would keep the Hgb threshold at 11 g/dl.
Donor Collection:
In the future, I would consider using one of the soon-to-be-released portable devices that continuously monitor vital signs with pO2 and EKG lead to rule out asymptomatic pulmonary or cardiac problems.
I would also consider using low-ABO-titer, group A, universally to meet the demand for group B and AB patients.
Donor Testing:
There is still no need to segregate and separately test CCP donor specimens from regular blood donor specimens. I would perform SARS-CoV-2 antibody testing and set a threshold for qualifying donors—that threshold will be based on the manufacturer’s recommendations. However, if the treating physician wanted to use a low-titer unit, I would permit this.
Donor Processing:
There is no need to change this from the current processes. Keep the CCP processing separate from the regular operations.
CCP Plasma Release:
I would keep the quarantine release and restrict it to the locations used for treating COVID-19 patients
Medinfo Software Modifications:
I would record the IgG and IgM titers for SARS-CoV-2 antibodies in each donation record. This would include testing and entering the results on donations prior to this testing. ISBT labels should include this antibody titer.
Hospital Information Software Modifications:
Set up restricted CCP ordering for the actual treating physicians only. Also provide the ISBT code and shortened descriptors to it if necessary (certain HIS vendors still cannot read ISBT codes natively).
The original CCP workflow is attached for reference.
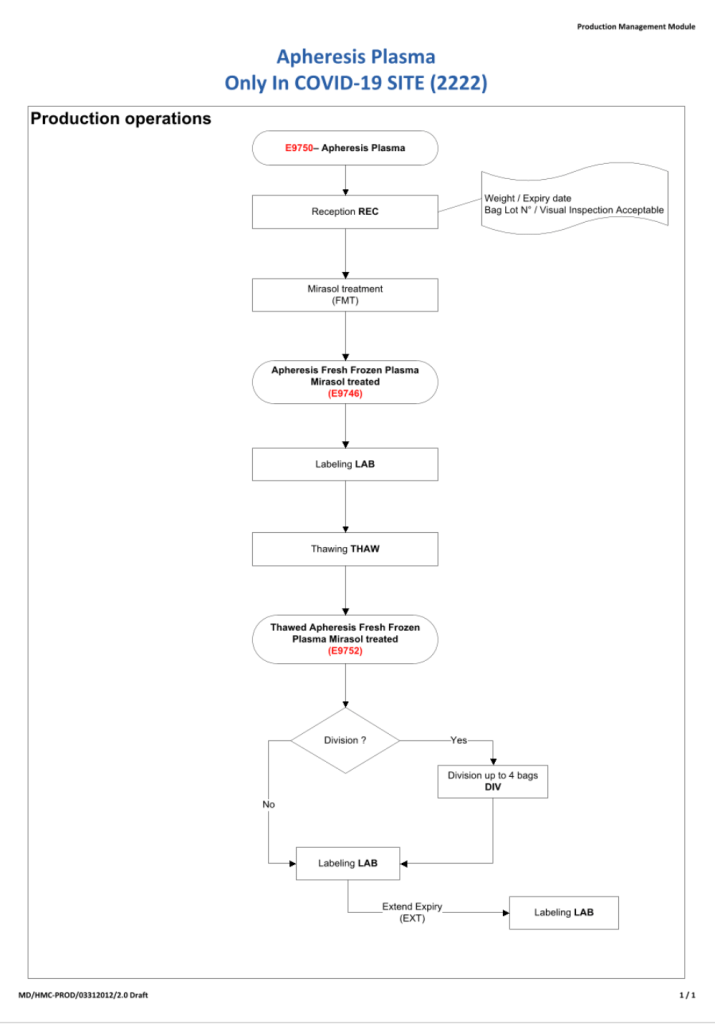
This process was originally done in the first phase of CCP collection. I have updated it to include SARS-CoV-2 antibody testing.
Principle:
Due to the pandemic, we will initially MANUALLY collect an experimental, investigational-use-only plasma product from apheresis donors and treat it with Mirasol. THIS IS A EMERGENCY INTERIM PROCESS UNTIL THE MEDINFO HEMATOS IIG PROCESSES ARE PREPARED AND VALIDATED.
Policy:
References:
All blood components are considered medications and are subject to Good Manufacturing Practices as mandated by international accreditation standards. The whole process must be done reproducibly and precisely by specific personnel trained and documented to be competent. This includes collection of convalescent COVID-19 plasma.
Transfusion Medicine will provide staff who are deemed competent for the entire process of the collection, manufacture, and release of this unlicensed, emergency-contingency component.
It will help greatly if all candidates are prescreened to exclude the following candidates:
This is NOT a complete list of criteria. Transfusion Medicine personnel will screen according to the full donor criteria. Thus, donors passing the pre-screening may still be otherwise disqualified based on the detailed process.
I am attaching the US Center for Biologics Evaluation and Research CBER Guidance for Industry revision dated December 2020 to replace the one issued in September 2019.
This is a very detailed document that will require US blood centers to comply with newer more stringent safeguards to minimize the risk of bacterial contamination of platelet components.
The easiest way to comply is to universally pathogen-inactivate all platelet components: then the rest of the algorithm does not apply. I am happy that for over 10 years I have used pathogen-inactivation (riboflavin-based Mirasol, Terumo BCT) and not experienced any bacterial sepsis from platelet or plasma components.
For those of us practicing outside the USA, please note:
The US still does not permit pooled, buffy coat platelets to have either a 5 or 7 day outdate. For pooled components stored at 20-24 C, the FDA only allows a four-hour outdate, regardless what the rest of the world permits. Thus, the USA mainly uses apheresis platelets.
If you have pathogen-inactivated platelets, you are so fortunate that you don’t have to follow these other recommendations to have a low risk of bacterial contamination.
Reference:
Bacterial Risk Control Strategies for Blood Collection Establishments and Transfusion Services to Enhance the Safety and Availability of Platelets for Transfusion, Guidance for Industry, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, September 2019 updated December 2020
CBER Guidance for Bacterial Contamination Guidance, Revised December 2020 (PDF)
