Principle:
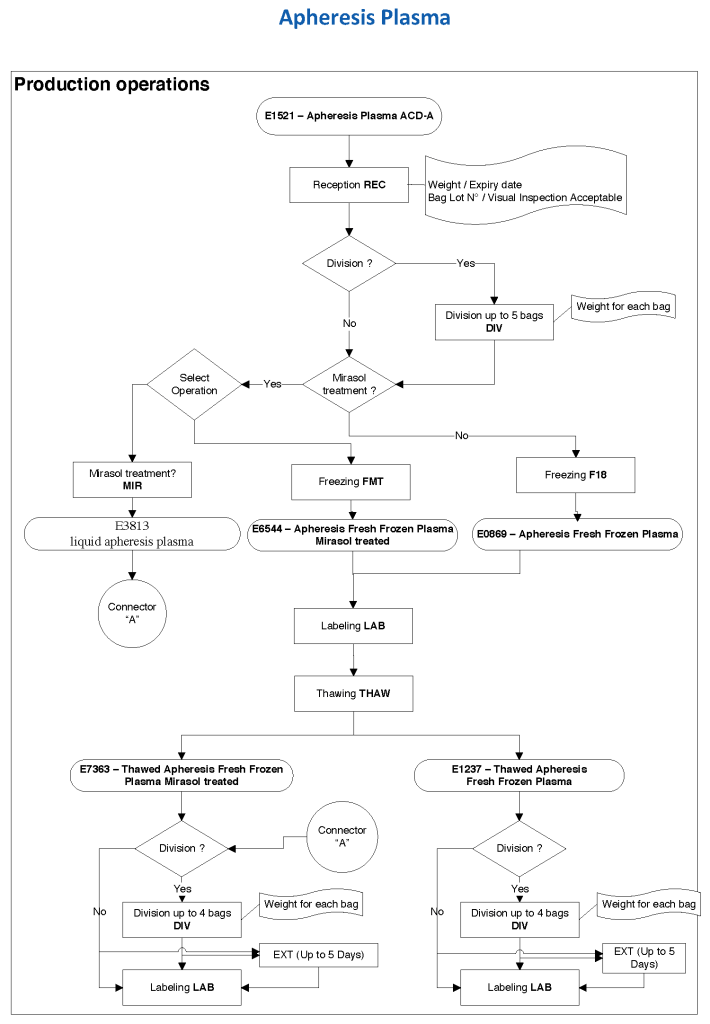
Plasma products (FFP, FP24, thawed plasma) are only available in limited quantities so wastage must be minimized. Thawed plasma has full factor activity for 24 hours; after 24 hours, all factors are still present at near normal levels except factors VIII and V. AABB Standards permit continued usage of thawed plasma stored between 1-6 Celsius as the component, “thawed plasma” for 5 days.
Thawed plasma may also be prepared directly at the time of production (i.e. without freezing) for certain cases (MTP, liver transplant protocol, plasma exchange) to shorten the release time. It is called liquid plasma since it was never frozen but should be considered equivalent to thawed FP24 if used within 24 hours or thawed plasma if used between 24 and 120 hours.
Liquid plasma, as we use here, is prepared directly from plasma treated with Mirasol and generally used within 5 days; however, in Medinfo HIIG, this plasma may have an outdate of 26 days in accordance with 21CFR610.53, but it is not used beyond five days except if approved by the Senior Consultant/Division Head of Transfusion Medicine. Since it is used within 5 days, it is equivalent to thawed plasma.
Definitions:
Responsible blood bank physician: specialist or consultant physician on-call at the time the discrepancy is detected
Policy Details:
- Plasma is dispensed without regard to the Rh(D) of the donor.
- Check if thawed plasma or liquid plasma (<5 days) is available first.
- Do not thaw plasma until the clinical service is ready to transfuse it.
- Tell clinical service to contact transfusion service 2-3 hours before intended transfusion time.
- Exceptions: liver transplant surgery, massive transfusion protocol, therapeutic apheresis, class 1 emergency requests
- If thawed plasma or liquid plasma is kept in the blood bank but not used:
- Reassign to another patient of compatible ABO type as thawed FFP/FFP24 if < 24 hours post-thaw or <24 hours of production if liquid plasma (see attached table).
- If thawed > 24 hours or liquid plasma between 24 and 120 hours post processing, reassign component as thawed plasma and use for up to 5 days. It is preferable to use thawed plasma < 24 hours old for neonates.
- Thawed >120 hours post-thawing and/or liquid plasma > 120 hours post-production should be discarded.
- Plasma usage and wastage will be monitored and reported to the Transfusion Committee.
Note:
Thawed plasma released from transfusion service should be discarded if returned.
References:
- Technical Manual, Current Edition, Bethesda, MD, USA
- Standards for Blood Banks and Transfusion Services Current Edition, AABB, Bethesda, MD, USA
- 21CFR610.531(c): Whole Blood and Blood Components Storage Temperatures and Dating Periods, Current Version
PERMISSIBLE FFP/FP24/THAWED PLASMA SUBSTITUTIONS
| PATIENT BLOOD TYPE | USE THE FOLLOWING: |
| O | ALL BLOOD TYPES |
| A | A or AB |
| B | B or AB |
| AB | AB |
24/10/20