I will be posting a detailed series about the manual and software-enhanced COVID-19 processes that I set up in Qatar at HMC Doha in March-April 2020.
In this series I will provide you with screen shots of my Medinfo Hematos IIG software design for each step in the process: collection, processing, testing, inter-depot transfer, and hospital transfusion service/blood bank release.
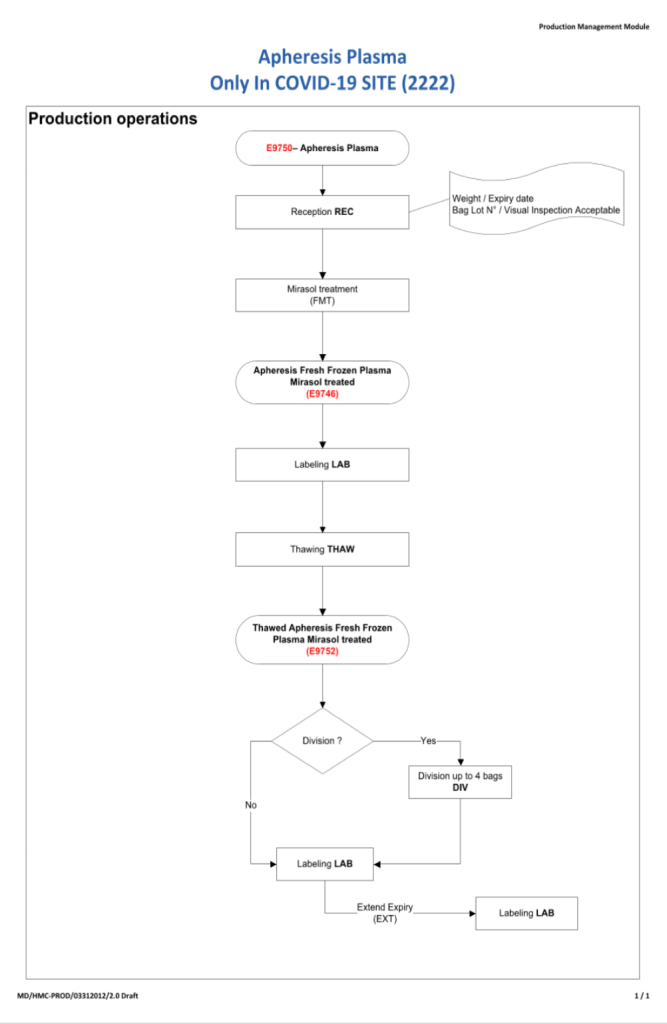
This GMP-compliant software-enhanced system is based on the manual system I set up in early March 2020 at HMC.
I want to thank Medinfo Hematos IIG for their rapid response to building this parallel system based on my standard processes in so short a time (two weeks) and my special thanks to the software engineering team at Vital Health Technologies, the agent for Medinfo in Qatar.
To start the series, I am providing the basic workflow for the system. As is normal in Medinfo software design, a full mapping of the processes are made. This workflow shows the new CCP ISBT codes and the quarantine collection and processing steps. The donor testing (marker and immunohematology) processes are similar to those for regular donor units.
This is basically the same process both manually and in the software. I always say:
A good software process is based on a good manual process!!
Please note the following workflow for our initial discussion.