This is a post of an old teaching presentation for pathology residents, hematology fellows, and transfusion medicine fellows from my time at NGHA-Riyadh.




















Teaching medical and technical staff transfusion medicine
This is a post of an old teaching presentation for pathology residents, hematology fellows, and transfusion medicine fellows from my time at NGHA-Riyadh.
The World Health Organization WHO just released a Key Facts document on HTLV-1 infection (references below) as follow up to their February, 2020 Technical Report. Here are some highlights for blood bankers:
HTLV-1 is efficiently transmitted by blood transfusion with a rate of 28-63% from a donor with HTLV-1 and up to 87% from a tissue transplant.
Testing can be made more complicated due to the length of time between contracting the virus andthe seroconversion required for the virus to appear on tests. This period has been reported to be aslong as 65 days.
Mandatory HTLV-1 antibody screening of all blood donations has been implemented in 23 countries.
Because HTLV-1 is almost always cell associated, leukoreduction may be as effective as blood donation screening in preventing transmission.
Following current practices, screening tests for HTLV-1 should be followed by confirmatory tests for the diagnosis of HTLV-1. Most screening tests use immunoassays, which rely on detecting anti-HTLV-1 antibodies. Commonly used confirmatory tests detect antibody responses to specific HTLV-1antigens. Test types include the Western Blot, radioimmunoprecipitation assay (RIA) and linear immunoblot assay LIA. However, the Western Blot test has been found to give unreliable results. Several studies have proposed transitioning from using Western Blot for confirmation in routine testing to using line immunoassay or NAT.
In my laboratory in Qatar, we detected approximately 8-10 cases of infection per year on a donor testing base of 36,000 for the year 2019. These were confirmed cases by LIA. In addition to the universal leukodepletion of all components to the CE-mandated level of < 1E6, we also pathogen-inactivated platelet components and plasma.
References:
This is a reprint of a previous post.



During my career, the role of the transfusion medicine physician in handling RBC antibodies has evolved. Depending on your location, either a specific transfusion medicine physician or a hematologist covering the specialty handled this. In the United States, most hematologists nowadays do not do this.
During my US residency training in pathology, most trainees did not have an interest in blood banking and often used the rotation to take vacations. For their board certification in clinical pathology, they crammed for the examination and afterwards did not engage in it.
When I started my career in the United States, hospitals had a hospital blood bank/transfusion service and handled most of their antibody problems themselves. In Chicago where I practiced, many blood banks had Specialist in Blood Banks SBB graduates or SBB students working, and of course, they had a dedicated blood bank supervisor.
Currently in the United States, the hospital blood banks may be staffed by generalists and there is no one with specific antibody experience. SBB graduates are expensive and usually work in blood centers or academic hospitals.
If you have a regional blood center with a reference laboratory, you can send your antibody workups there and let the blood center select appropriately antigen-matched units. The physician covering the blood bank does not have to get very involved.
In the Middle East in the systems that I have run, there have been no reference laboratories and often no regional blood center. The hospital system or the free-standing blood bank is an independent entity and must rely on itself. The physician responsible for the blood bank must review the antibody panels and make his own decision how to select RBC components.
In the Middle East, especially in the larger centers with academic hospitals, there is usually a transfusion medicine physician who reviews the antibody workups and makes the final decision of what RBC types to dispense. He does not have a reference laboratory to rely on. He is responsible for the choices and has no one to refer cases on a 24/7 basis.
In the Middle East, the transfusion medicine physician must be proactive. He must also select antigen-matched, fully or partially, and understand the trade-offs in cases with multiple antibodies. He must know how to deal with nonspecific antibodies and antibodies to high-incidence antigens, especially if he works in a region where the antibody panels are not optimized to the local population or there are many different ethnic groups in the local area.
Thus, training someone to practice in the Middle East requires spending considerable time in technical matters that might not be necessary if he/she practiced in the West. He may not only have to serve as a physician but he may be the equivalent of the reference laboratory supervisor. Until such time that reference laboratories are available, this model is essential for safe practice. Training programs for the region must reflect this and in particular, teach about specific antibodies common to the region. He must be technically oriented and he must take ultimate responsibility for the interpretation of the antibody workups and selection of the appropriately matched units.
This is updated version of a previous post.
This post is mainly on building processes for a non-turnkey system such as the Medinfo Hematos IIG software that I have worked with in several countries, but there will be a few words about turnkey systems for general laboratories.
This has been a collaborative effort between the software vendor’s engineers, my Super Users, and myself. This pluralistic approach has been most productive.
A turnkey system has pretty much already defined most of the basic processes—those have been specifically approved by a regulatory agency such as US FDA. There is little customization except formatting screen and reports. Instrument interfaces are also mainly predefined. This requires much less thought and planning than a custom-built system designed on the sites actual workflows, but it can be an exercise of putting a round peg in a square hole. You don’t always get what you want or need.
In the locations where I collaborated in setting up the Medinfo Hematos IIG program, we did not follow US FDA but mainly the Council of Europe CE standards since these were much more customizable. We could modify and add additional criteria specific to our country and region (e.g. rules for donor qualification for local pathogens). This has always been my preferred approach. Also, the USA does not use the full ISBT specification for its labels.
Start with a frame of reference (CE) and then try to optimize it for our local needs. Unfortunately for blood banking, FDA has many fewer approved options than other regions, including in the preparation of blood components, e.g. prohibiting the use of pooled buffy coat platelets, lack of automated blood component production such as Reveos, and use of world-class pathogen-inactivation technologies such as Mirasol.
If you invested the time to make a detailed workflow across all processes and tests, much of this can be readily translated into the software processes, but first you must study the flows and determine where you can optimize them. This requires that you study the options in the new software to see what you can use best.
I always liked Occam’s Razor, i.e. “ntia non sunt multiplicanda praeter necessitatem,”—the simpler the better as long as it meets your needs. If the manual processes are working well and can be translated into the new system, do so. If they need changes for optimization, then do so only if necessary.
Most of my career has been spent overseas with staff from many different countries and backgrounds, most of whom were not native in English. The wording of the processes is very important. Think of the additional obstacle of working with a complicated software in your non-mother tongue! Also consider the differences between American English, British English, and international English. I always made the Super Users read my proposed specifications and then asked them to repeat what I wrote/said. There were many surprises!
I think of the Aesop’s fable about the mother who gave birth to an ugly baby looking like a monkey. Still, to the mother her baby was the most beautiful baby and she entered him into a beauty contest. In other words, to the mother her child is perfect!
It is most important to use the manufacturer’s recommendations to build tests and for the special automated processing and pathogen-inactivation processes. For example, we had multiple ABO and D typing tests—they did not necessarily agree on what were acceptable results for automated release of results. The same is true for many other tests.
Example: One method for Rh(D) typing stated that only results in {0, 2+, 3+, 4+} were acceptable—all other results required manual review and/or additional testing. Another only accepted results in {0,3,4}. Thus we had to build separate D typing processes for each methodology.
Another consideration is whether to offer all the processes globally or restricted to one site. I favor allowing access to all methodologies at all sites—in case of a disaster where tests had to performed at another site. This means that if you send an order over an interface from the hospital system to the blood bank system, then at the receiving (blood bank) end, you would choose which methodology to use, i.e. it is not a one-to-one mapping but rather a one to many mapping.
If we changed equipment at one site to that used at another site, we didn’t have to modify our software to accommodate this. Even if you didn’t have the equipment or reagents at one site, you could always build it into the system and not activate the settings until needed.
Finally, the issue of middleware. Many instruments offer this, but one faces the problem about support and regression errors when you either update the middleware software or the blood bank computer software. Medinfo itself could serve as the middleware so there was less chance of errors when updating the software. In fact, I never used any middleware when using Medinfo.
This is a revised version of a previous post.
As much as 90% of the RBC component allocation can be performed without an actual crossmatch test (AHG or immediate-spin) provided that certain criteria are met.
Enforcing these rules, however, can be cumbersome unless one has blood bank software that verifies that each rule is met. In the Medinfo patient module, the transfusion history database is checked automatically. If the rules are met, then Medinfo allows the selection (allocation) of RBC units without performing a crossmatch test. Otherwise, it will check to see if the AHG crossmatch has been done within the past 3 days. If not, it will prompt for new crossmatch testing with a new specimen. If the situation is urgent, one can go to Emergency Mode and release components without the crossmatch.
Principle:
In selected patient categories, no classical crossmatch may be required for release of RBC components. The criteria are specified here as applicable in my build of the Medinfo Hematos IIG computer system HIIG.
Policy:
References:
This is an update of a previous post.
Principle:
In 1984 effective with the 13th Edition AABB Standards, the requirements for performing a direct antiglobulin test and autocontrol for compatibility testing were eliminated. The DAT is very important to detect delayed hemolytic transfusion reactions, certain autoimmune conditions, and drug-related hemolysis.
Since that time, the immediate-spin crossmatch and now the electronic computer paperless crossmatch may be used for most compatibility testing in place of the classic, antiglobulin-phase (indirect antiglobulin test) crossmatch.
If an antiglobulin phase (IAT) crossmatch is performed, an RBC unit with a positive DAT will cause a false-positive reaction. Since most crossmatching does not include the IAT, it will not be affected by the DAT status of a donor unit.
Policy:
Important: Don’t do a classic AHG/IAT phase crossmatch unless you have to do it (see conditions above.) A donor unit with a DAT is unlikely to be clinically significant and may be transfused safely to the patient in most situations. Patients receiving electronic-crossmatch and immediate-spin crossmatch are receiving units with positive DAT without incident.
References:
As a transfusion medicine physician, I was always being called to make decisions at all times when I was on-call. This could be for qualifying donors, therapeutic apheresis, or most commonly to analyze complex immunohematology situations and transfusion reactions.
I had to make decisions on data that was presented to me by my staff on the phone. I had to rely on the technical staff performing the test. If I made a decision on their findings and their findings were incorrect, I was still responsible for the error as head of the department.
Nowadays with a blood bank computer system, I could remotely access the data as it appeared in the computer. Still, how could I be sure that it was correct?
Based on my many years of experience, I have learned that I needed to know the capabilities of each staff member and to what extent I could rely on their results. Not everyone could do complex cases.
I also listened to how they presented the data to me. If it was disorganized or if they sounded uncertain, that was a red flag that something was wrong and I should be very careful about using the results. In such cases, I told the staff member to call a senior person to repeat the work. Of course, I did this in a delicate, face-savings way not to hurt the staff’s feeling. I usually told them to collect a new specimen and have a second senior person to repeat the workup.
I also had to know the context of the workload at the time of the consultation. Were there shortages of staff, were the staff stressed out, was there too much work for them to properly perform? In those situations, I would authorize additional staff to come in and repeat the work. I was very worried when outside hospital staff used to scream at my staff (usually junior doctors) and upset them. In the emotional distress, they could make a dangerous mistake. One of my roles was to serve as a counselor and de-stress my staff.
In summary, if you do not feel your staff can handle the work properly, don’t rely on the output. Repeat the work, defuse the stress, fix the workload, etc. As the transfusion medicine physician, it is also your neck on the line. You are responsible to determine if you can trust the results to make a medical judgment.
This is an update of a previous post.
I have been involved with planning for several plasma fractionation projects in the Middle East.
Many clients expressed the interest in using local plasma to make plasma derivatives (e.g. factor concentrates, intravenous gamma globulin, albumin), feeling that local plasma was safer than using imported plasma. Some of these are in short supply in the world market so the only way to ensure their uninterrupted availability is to consider to manufacture them for local consumption.
Still, the major issue today is that it is difficult for any country in the region to collect enough plasma to make such a project feasible. When I first considered such planning, we were looking for as much as 250,000 raw liters of plasma annually. Since then, there are newer technologies that allow much smaller batches to be cost-effective. Alternatively, one could charge higher prices for using smaller batches from local plasma.
Still, it is likely that plasma must be imported to sustain a plant. There are different regulations for plasma donor qualification country-to-country. Many of these jurisdictions may do less screening and testing than is done for normal blood and apheresis donors. Other countries use their blood donors with the same requirement for both commercial plasma and blood donations.
In this era of emerging infectious diseases, I personally favor using the stringent blood donor criteria—same as routine collections. It is not what we know, but the unknown pathogens that are potentially the most dangerous.
In addition to building a fractionation plant, one must train staff for this highly technical operation. This may require developing a special curriculum to prepare students for these jobs.
To export the plasma to certain regions, one may have to use plasma quarantine. In this protocol, plasma is held or quarantined until the next donation is collected and passes screening. This requires a robust blood bank production software such as Medinfo to track serial donations.
There are other processes to consider: how to develop a transport network to keep plasma frozen at minus 80C viable in a region that reaches very high ambient temperatures.
I would recommend a graded approach to develop such an industry. First I would negotiate a plasma self-sufficiency arrangement. We would collect local plasma in the country and export it to a manufacturing plant in another country and the derivatives would be returned to us. This may require inspection by the accreditation agency of the processing country to allow importation of the raw plasma for manufacture.
Since it is unlikely any one country has enough plasma for manufacture, recruiting neighboring countries to participate in a manufacturing plant is important. Technology for such a plant is complex so establishing a joint venture with one of the plasma industry companies is essential. Some manufacturers are very keen to develop extra capacity since there is a world-wide shortage of plasma fractionation and are even willing to help obtain external plasma sources for such a plant.
Such a plant is an excellent way to develop local talent to run such a plant, including training of local staff to be the industrial engineers in the plasma fractionation process. It would take approximately two years of training to prepare engineers on-site at a plasma fractionation site if they have studied the necessary science and mathematics subjects.
Such a program would take several years of planning and development. Some of the major steps needed include:
When a new software version was introduced in my system, first the vendor did a preliminary round of testing before submitting it to my Medinfo-Laboratory Information System team for validation. I then prepared a training session for the Super Users and assigned them validation tasks. When the validation was completed and accepted by me, then the software was submitted to the Hospital Information System HIS department, which conducted its own final acceptance training. Following this, transfusion medicine staff were trained before the new software went live.
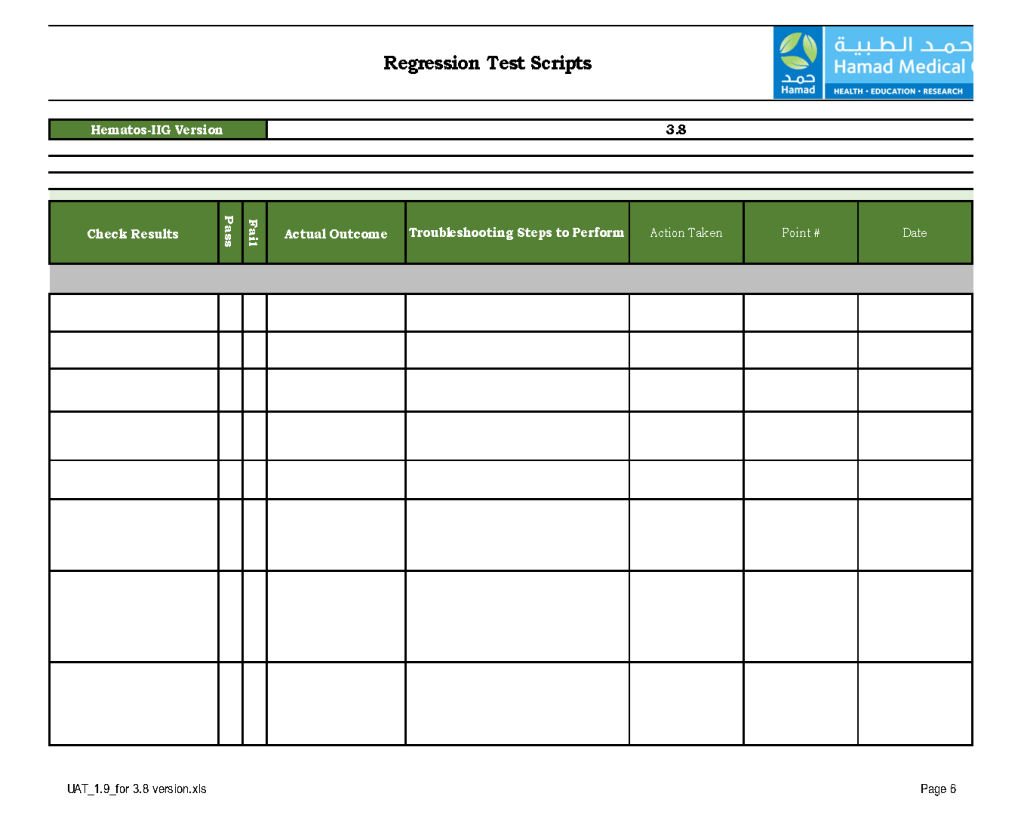
This is a sample user acceptance training document, which was prepared by Medinfo and myself and submitted to HIS for the upgrade from Version 3.8 to 5.0. The patient module is shown.
The Medinfo Super Users performed the script while the HIS Quality Team viewed the actual output. Notice for each Action step there is the Expected Result.
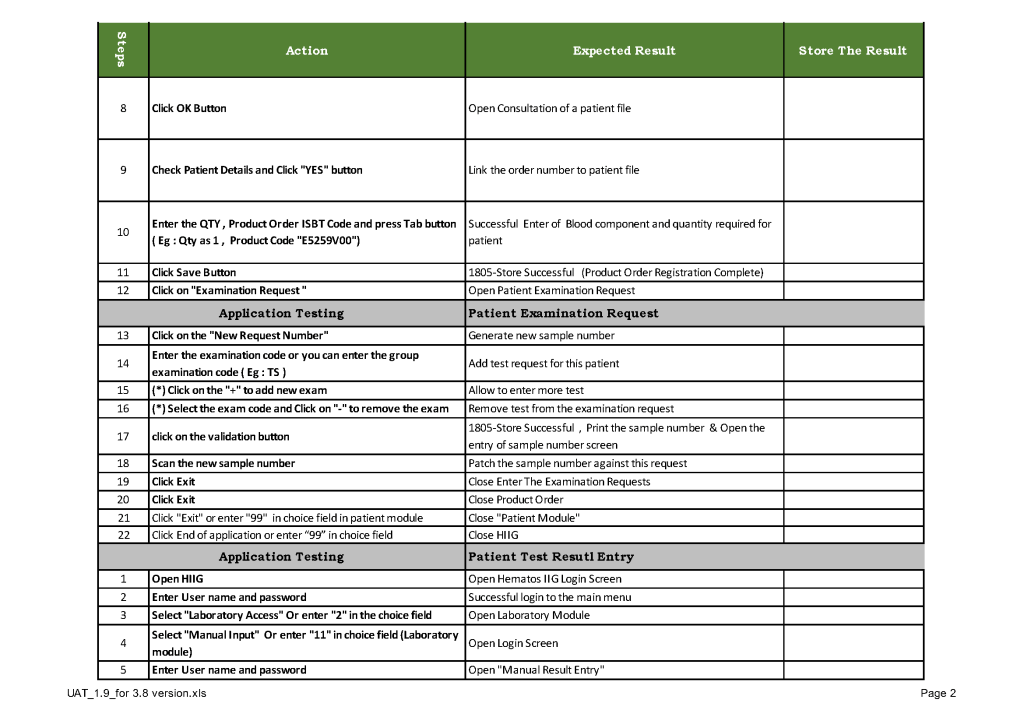
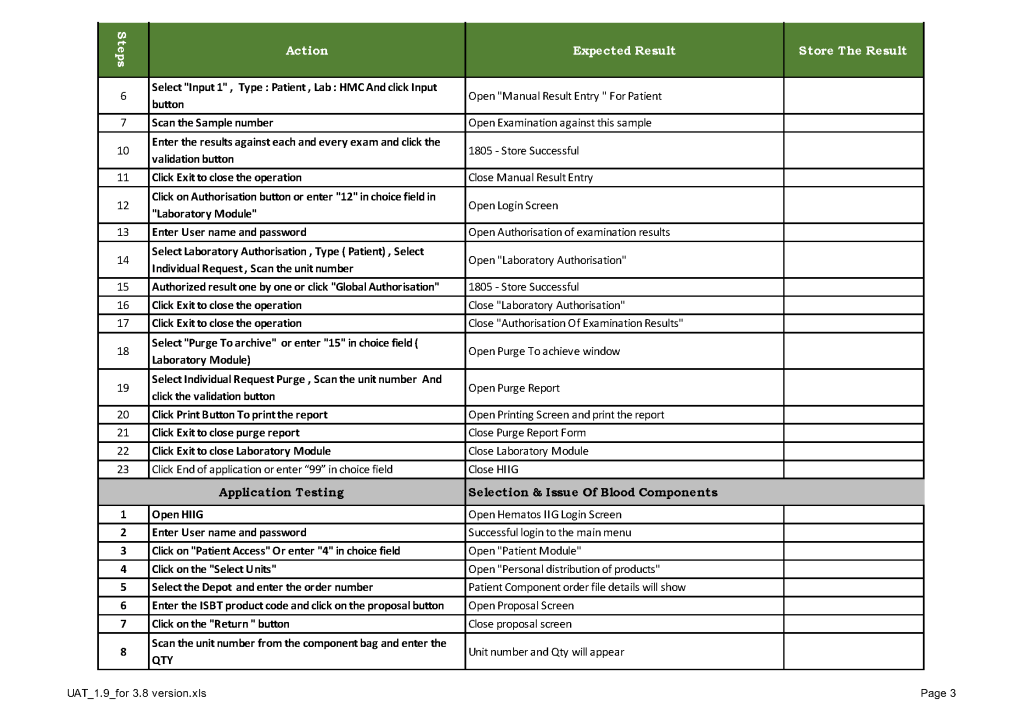
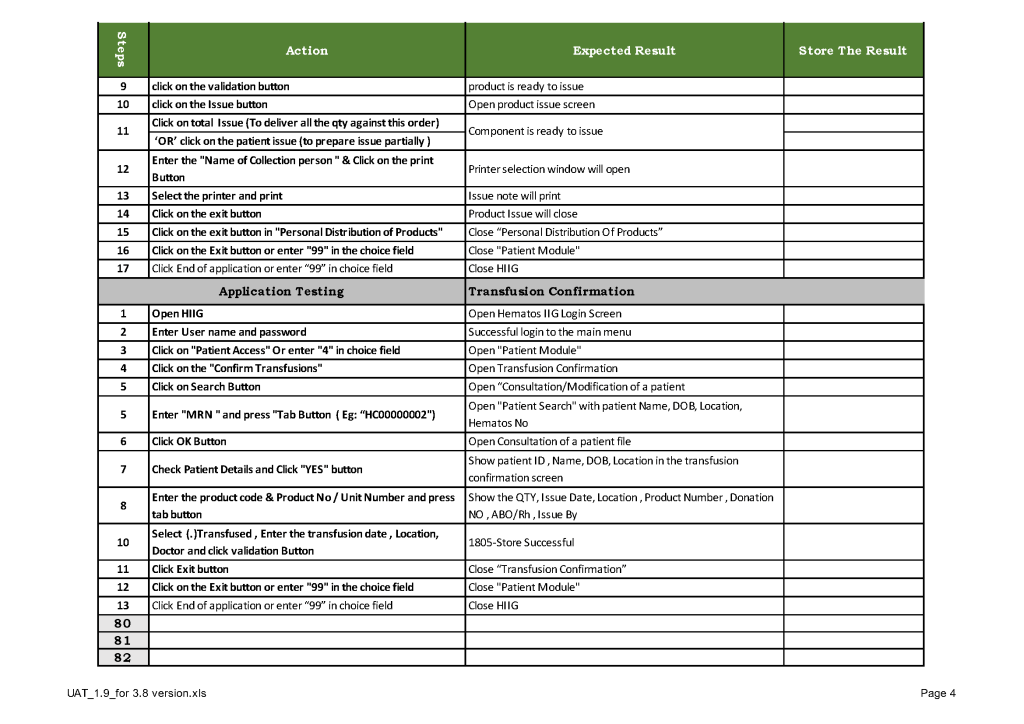
The evaluation for each step was recorded as part of this long and wide spreadsheet: