There are many variations for this most important test. In the very least, this consists of:
Screening polyspecific AHG for detecting IgG and/or complement
If positive, reflex monospecific tests to distinguish IgG from C3 reactivity
There are many kinds of reagents:
Immunoglobulin: whole molecule IgG, monospecific gamma heavy chain, monospecific alpha heavy chain, monospecific mu heavy chain
Complement: C3d, C3c, C3b, C5, etc
Regretfully, some of the monospecific are not really monospecific (e.g. whole molecule IgG detects both heavy and light chains and thus may detect IgG and give weaker results for IgM or even IgA.)
In 1984 effective with the 13th Edition AABB Standards, the requirements for performing a direct antiglobulin test and autocontrol for compatibility testing were eliminated. The DAT is very important to detect delayed hemolytic transfusion reactions, certain autoimmune conditions, and drug-related hemolysis.
Since that time, the immediate-spin crossmatch and now the electronic computer paperless crossmatch may be used for most compatibility testing in place of the classic, antiglobulin-phase (indirect antiglobulin test) crossmatch.
If an antiglobulin phase (indirect antiglobulin test–IAT) crossmatch is performed, a donor unit with a positive DAT will cause a false-positive reaction. Since most crossmatching does not include the IAT, it will not be affected by the DAT status of a donor unit.
Policy:
Donor RBC units will NOT be routinely tested for DAT as part of component processing.
The type of compatibility testing selected for a particular patient should be the technically simplest one (no need to do extra work unless so instructed by the transfusion medicine consultant/designate):
Do a full antiglobulin-phase IAT crossmatch if ANY of the following applies:
There are no two independent ABO/D typings on the patient during the current admission.
The ABO/D type of the current admission does not match the historical information.
The patient has a detectable antibody at 37C
The patient has a history of a clinically significant antibody but no current antibody
Whenever the consultant, transfusion medicine/designate requests it.
Whenever the Medinfo HIIG record so indicates (in comment section)
Do the immediate-spin crossmatch if ALL of the following apply:
Only one determination of the ABO/D type
The historical ABO/D type agrees with the current type.
There are no antibodies reacting at 37C AND there is no history of antibodies at 37C.
Use the computer/electronic crossmatch if ALL of the following apply:
There are two determinations of the ABO/D type and they both agree with each other.
The historical ABO/D type agrees with the current type.
There are no antibodies reacting at 37C AND there is no history of antibodies at 37C.
When to do a DAT on a donor unit:
Patient antibody screen is negative but the full AHG crossmatch is incompatible.
Part of a transfusion reaction workup where the AHG crossmatch of donor cells and patient serum is incompatible.
Whenever the consultant, transfusion medicine/designate requests it.
If a donor unit is found with a positive DAT, perform:
Do polyspecific and monospecific (IgG and C3) antisera
Perform an acid-elution.
Send the results to the transfusion medicine consultant/designate for review.
The reviewer will enter his review in the Medinfo HIIG in the Donor Consultation Section both as global donor comment and a result-specific comment against the antibody screen result.
Use of the DAT-positive donor unit:
Select another RBC unit for the transfusion.
The final decision to use the unit will be made by the Transfusion Medicine consultant/designate.
Important: Don’t do a classic AHG/IAT phase crossmatch unless you have to do it (see conditions above.) A donor unit with a DAT is unlikely to be clinically significant and may be transfused safely to the patient in most situations. Patients receiving electronic-crossmatch and immediate-spin crossmatch are receiving units with positive DAT without incident.
References:
Standards for Blood Banks and Transfusion Services, Current Edition, AABB, Bethesda, MD, USA
Guidelines to the Preparation, Use, and Quality Assurance of Blood Components, European Committee (Partial Agreement) on Blood Transfusion (CD-P-TS), Current Edition
Technical Manual, Current Edition, AABB, Bethesda, MD, USA
This is the process I developed for HMC Doha. The Medical Director (here Head, Transfusion Medicine HTM) is actively involved in the development of policies, processes, and procedures for ALL types of autologous donation in conjunction with the National Transfusion Committee NTC.
Predeposit: Directly under the control of the HTM for all aspects: policies, procedures, and direct performance of the procedures, including annual review of criteria
Perioperative: HTM involved in conjunction with Surgery and Anesthesia through the NTC.
Intraoperative: HTM involved in conjunction with Surgery and Anesthesia through the NTC.
Postoperative: HTM involved in conjunction with Surgery and Anesthesia through the NTC.
Background:
There are four basic types of autologous transfusion: preoperative, perioperative hemodilution, intraoperative, and postoperative drainage/collection. The use of all of the above techniques can significantly decrease the need for homologous blood and as an added benefit reduce the risk of the disease transmission and immunosuppressive effects of such homologous transfusions.
Preoperative collection can make available packed red blood cells, whole blood, platelets, FFP, and/or cryoprecipitate. However, at most two units of blood per week can be collected. RBC’s can be stored for up to 42 days in the liquid state, frozen RBC’s up to ten years, platelets up to five days, and fresh frozen plasma and cryoprecipitate up to one year. The last collection cannot be less than 72 hours prior to the surgery time. Units can be collected as long as the patient’s hematocrit remains above 33%. Supplemental iron and erythropoietin can increase the number of units harvested. The biggest obstacle to using this service is the coordination of the patient scheduling for this procedure. The blood bank does not have the resources to prospectively analyze the surgical scheduling and make the various appointments, contact the attending physician, etc. Thus, this service is vastly underutilized.
PHD or Perioperative hemodilution (also called acute normovolemic hemodilution) is useful in cases when the anticipated blood loss is at least one liter and the initial hematocrit is at least 34%. This includes essentially all types of surgery, but in particular cardiac, vascular, orthopedic, and urologic cases. The patient’s hematocrit Hct. is lowered to the range of 20-25% and the blood is replaced by crystalloid in a ratio of 3:1–i.e. three times as much fluid as blood, or in the case of colloid replacement, a 1:1 ratio of colloid plus 0.5 to 1.0 ml. of crystalloid. Crystalloid has the advantage of being readily removed by diuretic use. However, this technique should not be undertaken when vascular access is inadequate or appropriate monitoring devices are lacking. The physician performing PHD must be familiar with the compensatory mechanisms normally invoked when the hemoglobin is acutely lowered.
Another new twist to PHD is the perioperative collection of platelets by a special attachment to a cell-saving machine. This could allow collection of a typical apheresis load, about 6 to 10 units of fresh platelets for potential use. There are currently studies underway to determine if this has particular clinical advantages to warrant the additional cost.
Intraoperative salvage may be performed with a number of canister or automated devices. The latter is usually used when there are large volumes (usually 3 or more units) of blood to be salvaged. Depending on the body site, the recovered material is at least filtered and may or may not be washed. Care must be taken to collect the blood at a low suction rate and with minimal turbulence to minimize hemolysis.
Postoperative drainage collection of certain sites such as post-knee replacement surgery or chest wounds involves a canister collection device. This blood may or may not be filtered before reinfusion.
Note that perioperative and intraoperative material can only be transfused up to six or eight hours at room temperature or 24 hours if refrigerated at 1-6 degrees (depending on the method used) post collection to minimize the risk of infection. Intraoperative collection is usually contraindicated in cases of cancer and if the bowel has been violated.
Other Issues:
The transfusion criteria for autologous blood is the same as for allogeneic units. If you wouldn’t transfuse if no autologous blood were available, you shouldn’t transfuse because you have it!
The same compatibility testing algorithm applies both the autologous and allogeneic units.
Policy:
Scope:
Predeposit collection of Whole Blood/RBCs and plasma is under the authority of Transfusion Medicine.
Perioperative hemodilution, intraoperative cell salvage, and postoperative drainage collection is under the authority of the National Transfusion Committee in conjunction with the Departments of Surgery and Anesthesia.
Head, Transfusion Medicine will liaise with the clinical departments as needed.
Transfusion Medicine may provide blood bags for perioperative hemodilution upon request.
Transfusion Medicine does not receive autologous collections—perioperative, intraoperative, or postoperative drainage collection.
Processes directly under Transfusion Medicine authority:
Autologous collection of whole blood/RBCs (predeposit) for elective surgeries will be considered especially if the patient has a dangerous antibody for which antigen-matched units cannot be easily obtained (e.g. anti-k (cellano), anti-PP1Pk (anti-Tja), anti-H (Bombay and Para-Bombay phenotypes).
Autologous collection of plasma may be considered for patients with IgA deficiency with documented specific anti-IgA antibodies.
Autologous collection of platelets may be considered for patients with anti-platelet antibodies and platelet refractoriness.
Other requests will be reviewed by the Head, Transfusion Medicine or designate.
The final decision to proceed with items 2.1 and 2.2 will be made by the Head, Transfusion Medicine or his designate.
Process for Transfusion Medicine Autologous Procedures
The requesting physician shall provide a written order to the Blood Donor Center.
The request will be reviewed by a transfusion medicine physician.
If rejected, the requesting physician will be notified with the reason for the rejection.
If approved, the donor shall be screened by the usual donation process except:
Hgb >= 11 g/dl will be acceptable for whole blood collection.
Females may also donate autologous plasma.
The last autologous donation will be at least 72 hours before the elective procedure.
Marker testing: Components from autologous donors with confirmed positive cases of HBV, HCV, HIV, syphilis, malaria, or HTLV infection will NOT be used for autologous donation and will be destroyed.
Computer: Autologous collections will be entered as specifically as such in the Medinfo Hematos IIG blood bank computer system and be labeled as autologous in their corresponding ISBT labels.
Crossover of autologous units requires review and approval by a transfusion medicine physician–only those cases where the donor met the standard donor criteria will be considered.
The transfusion criteria for autologous units shall be the same as for homologous blood.
Both autologous and allogeneic units will follow the same compatibility testing algorithm.
References:
Standards for Blood Banks and Transfusion Services, 29th Edition, AABB, Bethesda, MD, USA, 2014
The electronic/computer crossmatch is vital to the hospital blood bank’s operation. It allows us to reduce unnecessary work and redeploy a very limited, precious workforce. As demonstrated in the previous post, it has many rules for current and previous testing and historical transfusion records.
This is NOT a one-time validation: it must be repeated to check for regression errors when updating the software version, regardless if there are any changes to its logic. It required many Super-Users to perform:
A test environment distinct from the live was used.
Patients had to be created and specific test results entered to match the algorithm below.
Donors had to be created and components made (including testing, production, release, inter-depot transfer)
Hospital blood bank had to receive the created units and enter them into active stock (and repeat the ABO/D testing to do this)
This is a sample validation plan created by me and used at HMC Doha for the Medinfo Hematos IIG software:
2 ABO/D determinations, one current within 72 hours
Second determination from history or second current sample
No ABO/D typing discrepancies
Negative antibody screen
No history of antibodies
40 patients with contraindications to electronic crossmatch:
10 with history of antibody
10 with current antibody
10 without 2 ABO/D determinations
10 with emergency release
Try to release RBC unit without further testing in each case.
Super Users had to capture screen shots of the transactions and send them to me as Division Head, Transfusion Medicine/LIS for review and acceptance.
Criteria for Acceptability:
All tested processes must complete without error, including recognition of ABO/D discrepancies as such.
System only allows computer release (electronic crossmatch) in those cases meeting computer-crossmatch criteria as shown in the following interim policy.
INTERIM POLICY: ELECTRONIC (COMPUTER) CROSSMATCH
Revised Date: 1/2/18
Principle:
In selected patient categories, no classical crossmatch may be required for release of RBC components. The criteria are specified here as applicable in our Medinfo Hematos IIG computer system HIIG.
Policy:
An electronic crossmatch without antiglobulin or immediate-spin phase testing may be used for the following patient categories:
The current ABO/D type matches the historical ABO/D type.
The ABO/D type (forward and reverse) is clearly defined without any discrepancies.
Two determination of the ABO/D group must be made:
One from a current specimen (within the past 72 hours)
The second by one of the following methods:
Testing a second current specimen
Comparison with previous records of ABO/D typing
Retesting the same specimen
The current antibody screen is negative
There is no history of RBC antibodies or a non-negative antibody screen.
General computer system safeguards:
The system contains the donor unit number, component name, confirmed ABO/D typing, two unique recipient identifiers, recipient ABO/D, antibody screen typing, and interpretation of compatibility
A method exists to verify correct entry of data before release of blood or blood components.
The system contains logic to alert the user to discrepancies between donor ABO/D group on the unit label and those determined by blood confirmatory tests and to ABO incompatibility between the recipient and donor unit.
HIIG will enforce the above rules.
References:
HIIG Workflow 1004, Patient Testing, 2013
Section 5.16, Standards for Blood Banks and Transfusion Services, Current Edition, AABB, Bethesda, MD, USA
FDA Guidance for Industry: Computer Crossmatch (Computer Analysis of the Compatibility between the Donor’s Cell Type and the Recipient’s Serum or Plasma Type), April, 2011
As much as 90% of the RBC component allocation can be performed without an actual crossmatch test—AHG or immediate-spin provided that certain criteria are met.
Enforcing these rules, however, can be cumbersome unless one has blood bank software that verifies that each rule is met. In the Medinfo patient module, the transfusion history database is checked automatically. If the rules are met, then Medinfo allows the selection (allocation) of RBC units without performing a crossmatch test. Otherwise, it will check to see if the AHG crossmatch has been done within the past 3 days. If not, it will prompt for new crossmatch testing with a new specimen. If the situation is urgent, one can go to Emergency Mode and release components without the crossmatch.
The following is the latest document I prepared on this process before leaving HMC. Please note that there was an extensive validation before this was activated for patient use.
INTERIM POLICY: ELECTRONIC (COMPUTER) CROSSMATCH
Revised Date: 1/2/18
Principle:
In selected patient categories, no classical crossmatch may be required for release of RBC components. The criteria are specified here as applicable in our Medinfo Hematos IIG computer system HIIG.
Policy:
An electronic crossmatch without antiglobulin or immediate-spin phase testing may be used for the following patient categories:
The current ABO/D type matches the historical ABO/D type.
The ABO/D type (forward and reverse) is clearly defined without any discrepancies.
Two determination of the ABO/D group must be made:
One from a current specimen (within the past 72 hours)
The second by one of the following methods:
Testing a second current specimen
Comparison with previous records of ABO/D typing
Retesting the same specimen
The current antibody screen is negative
There is no history of RBC antibodies or a non-negative antibody screen.
General computer system safeguards:
The system contains the donor unit number, component name, confirmed ABO/D typing, two unique recipient identifiers, recipient ABO/D, antibody screen typing, and interpretation of compatibility
A method exists to verify correct entry of data before release of blood or blood components.
The system contains logic to alert the user to discrepancies between donor ABO/D group on the unit label and those determined by blood confirmatory tests and to ABO incompatibility between the recipient and donor unit.
HIIG will enforce the above rules.
References:
HIIG Workflow 1004, Patient Testing, 2013
Section 5.16, Standards for Blood Banks and Transfusion Services, Current Edition, AABB, Bethesda, MD, USA
FDA Guidance for Industry: Computer Crossmatch (Computer Analysis of the Compatibility between the Donor’s Cell Type and the Recipient’s Serum or Plasma Type), April, 2011
I try never to forget that in most cases, the simplest solution is the best. This applies to software as well. The ideal situation is to have one interface to the blood bank software.
In many laboratory softwares, there is a middleware to interpret the raw data as it comes out of the equipment which may reformat it and interpret it. This approach means that you have to have one interface then middleware and then finally the software.
Inevitably, both the middleware and the laboratory software will need updating. Can you assume that they will still work after either or both are updated? Will there be regression errors?
A good example of regression errors is the Microsoft Windows Feature Update (sometimes referred to as the Update from Hell!). Previously working functionality gets broken, data can be lost, etc.
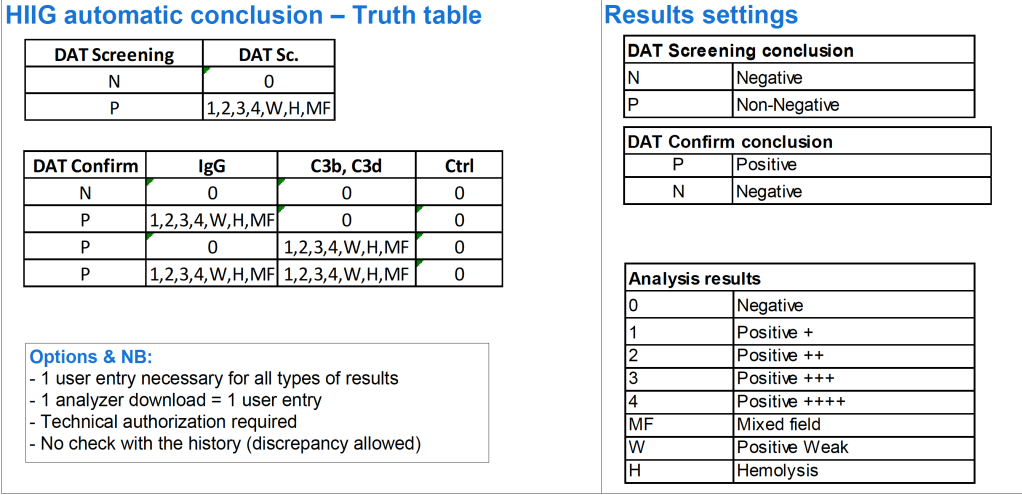
In Medinfo, you store a truth table of possible results and interpretations. Medinfo will directly read the machine interface data, interpret it according to the rules you make. The data can be numeric or alphanumeric or both. Each test, each equipment can have its own unique rules if necessary.
In my opinion, it is best to avoid middleware if your blood bank software can perform this function directly. When you upgrade it, it is much less likely to show errors—and you only have to deal with one vendor. Can you be certain that the middleware vendor and the blood bank software vendor will work well together to resolve any issues?
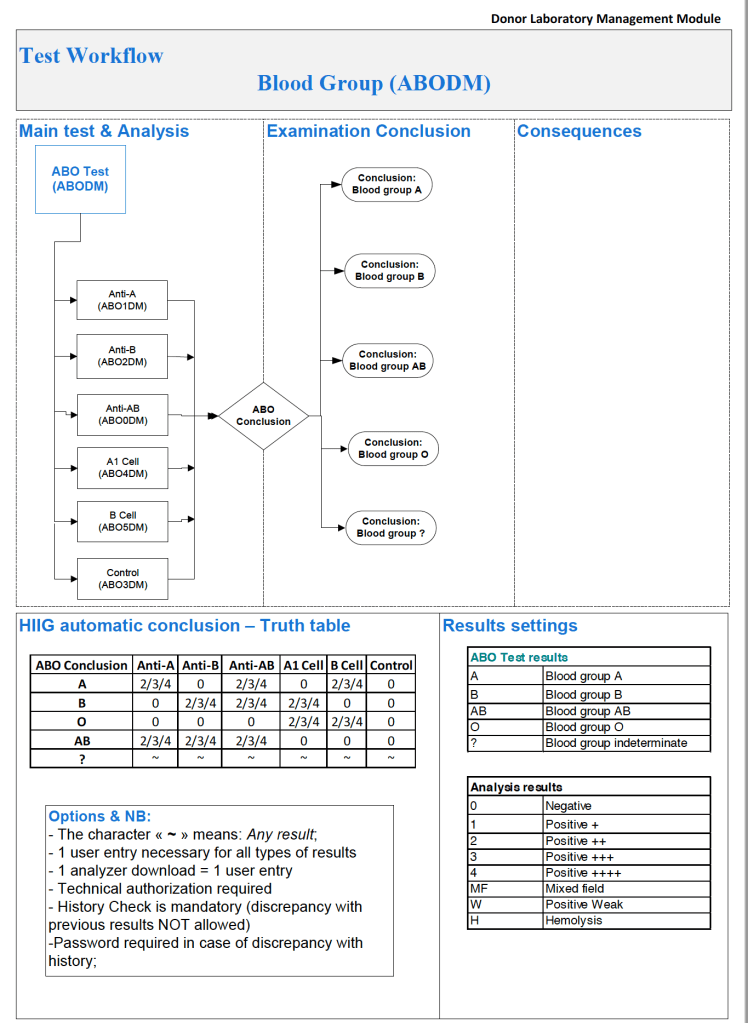
The following are some examples of truth tables, i.e. the rules for interpretation and disposition of interface results actually used at HMC Doha during my tenure there:
Example 1: Blood Component Production Truth Table
Production can only proceed if the volumes are within the specified ranges. This is very important if you are going to perform pathogen-inactivation since the ultraviolet illuminator requires a specific range, even more so if you are adding Mirasol (riboflavin) and PAS (platelet additive solution).
Example 2: ABO/D Antigen Typing:
One manufacturer’s requirements:
Example 3: Complicated D Typing Algorithm:
The acceptable range for automatic interpretation is much more complicated for D typing with the Ortho Vision MAX and uses 3 different monoclonal cocktails:
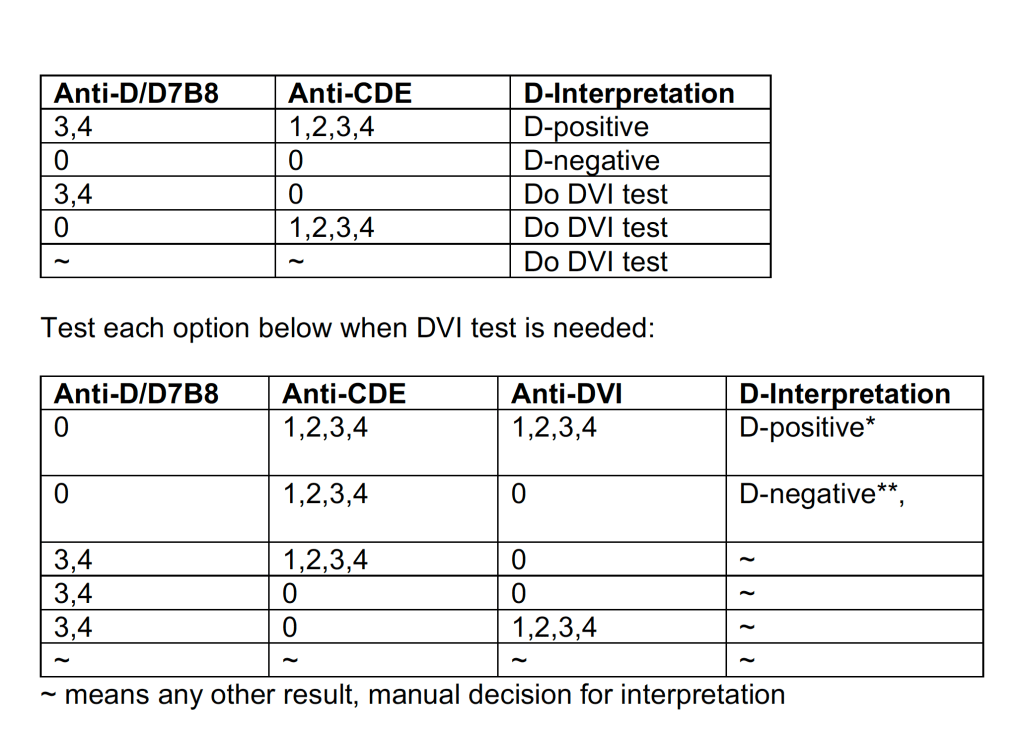
Example 4: Direct Antiglobulin Test Algorithm:
Both alphanumeric and numeric results are used.
Example 5: Four-Cell Antibody Screen:
Truth table for interpretation requires both alphanumeric and numeric results:
Conclusion:
If you are fortunate to have a dedicated blood bank software, you may not need middleware. Otherwise, you may need to use it for linking to general laboratory systems. Hopefully, your vendors will cooperate with each other.
The following post is based on my experience 2011-2020 at HMC through 16/4/20:
I have had many posts about the Blood Donor Center from registration, collection, processing, testing, and dispatch (inter-depot transfer). The hospital transfusion service or hospital blood bank continues the process on selection of the appropriate blood component for the patient. Specifically, it:
Verifies the ABO/D type of RBCs and ABO type of plasma components received
Physically examines each unit checking for leaks, labelling errors, etc.
Receives into stock the various components
Performs basic type and screen (group and save) testing of the patient including ABO/D type and antibody screen
Identifies antibodies if the antibody screen is non-negative
Performs direct antiglobulin test and elution if positive
Modifies components (thawing, aliquoting, irradiating, washing, pooling)—although in some sites, these latter functions may be performed in the Blood Donor Center
Performs compatibility testing and selects the appropriate method (electronic, immediate-spin, antiglobulin phase crossmatch)
Releases blood components to outside staff (nurses, doctors, etc. as allowed by the local authority)
Investigates transfusion reactions
When I was at HMC which included many hospital blood banks, we standardizes our methodologies/processes as much as possible, but we still had some differences based on the equipment at each site. When we built the blood bank computer system, we had to build a specific process for each test, taking into account the methodology and the type of reagents used. We used the manufacturer’s recommendations when establishing the criteria for each test.
There were manual and multiple automated tests, e.g. for ABO/D typing. Rules were established when automated release was allowed and when a manual review was necessary. Complicated cases were referred to the transfusion medicine physician for review and comment.
In our system, all tests could be ordered and performed from all sites. Transfusion medicine physicians could review all work from all sites. For technologists, they were restricted to the sites they worked or supervised except to review results.
All patient results across the entire system from the current and previous system were available and could be used to make/enforce rules.
In general, certain categories of results were referred to the transfusion medicine physician for review, but any test could reviewed by him/her, especially if a clinician requested it. Everything was documented in the software.
Component modification (thawing, aliquoting, irradiating, pooling, washing) processes were the same at all sites AND the blood donor center. Each modification changed the ISBT designation of the component, a new ISBT label was printed, and the outdate of the components were updated.
Antibody workups were still performed manually, but direct antiglobulin tests could be manual or automated. In each case, review with an interpretative comment was made by the transfusion medicine physicians and might include recommendations for selection and use of components. Rules enforced by the software could be made to enforce these recommendations.
This Powerpoint file summarizes the past previous posts about the use of automated components, pathogen inactivation, and their synergism with the blood bank computer software Medinfo Hematos IIG
Blood Bank instruments may perform tests and release test results in a numerical or alphanumeric format or both. For example, nucleic acid and enzyme immunoassay may release a qualitative result (e.g. positive, reactive, borderline/grayzone, negative, nonreactive). Alternatively, the machine may release the signal to cutoff ratio (S/CO) as a numeric result.
Blood bank software may use either kind of result on which to base interpretative rules for acceptability of the donor. The qualitative result criteria are based on the quantitative SC/O but the equipment automatically interprets this. The S/CO ratio of 1 is the cut-off point. Thus a value of 0.99 is negative and the value 1.01 is positive. But is it really so clear-cut since the difference between the two is so small? Thus, some people have added the term grayzone for values close to but below the cutoff. Could a value of 0.95 be an early infection?
I personally prefer to see the actual cutoff but use the manufacturer’s criteria for interpretation. As a physician, it is good to review the S/CO on serial exams. If a borderline or grayzone result becomes positive, then perhaps the original result indicated early infection. The question still remains, what is the grayzone? 0.95 to 0.99, 0.90 to 0.99, etc. Some accrediting schema have not used grayzone for interpretation.
With Medinfo’s blood bank software, I could chose either option or both—or at least store the S/CO as a nonreported result for subsequent review. I could even chose, test by test, in a series between reporting either S/CO or the qualitative result.
Semiquantitative results, e.g. in {0, 1+, 2+, 3+, 4+} are qualitative and could also include mixed field (mf) and hemolyzed (h). I showed examples of this with ABO/D antigen typing in a previous post—see attachment.
On the contrary, the results from blood production equipment may include parameters such as time of preparation, original volume, final volumes for each component, platelet yield index as an indirect measure of platelet count. When there is pooling, the final total volume is critical to determine if pathogen-inactivation procedures and platelet additive solution can be used. This is a much more complicated interface.
The blood production equipment interface issues will be considered in a future post.
When buying equipment while planning/implementing new laboratory software, I originally had a rule not to purchase anything that the vendor did not have a ready interface. Even that was not so clear since some vendors had interfaces listed as alpha, beta, and completed.
Could you use an alpha or beta interface? Was it safe for patient care? What was the development cycle for new interfaces with your vendor—months, years?
Even if the vendor had a completed interface? How “complete” was it? Did it accept all data from the machine? Did the data stream require reformatting? Who would write the transformational script?
Even if the vendor could support it, could your local IT organization and the local agent’s IT staff do it? I had plenty of headaches over this. The best equipment with the best interface that the local agent could not support was worthless to me.
Some finished interfaces took months to install because of connectivity issues? What version of the operating system was used? Was it secure? Did our IT department accept that version (e.g. Windows 7) and the provided malware protection? I have seen malware spread across a network from the interface software installed by the vendor, threatening the entire corporate system.
Did the solution require middleware? What were the implications of having middleware and its affect on the main software program, especially at the time of its upgrade or the main software?
I have seen vendors using Windows 2000 for their interface software as late as 2017. It was difficult for some of them to update to current, more secure versions. Anyway, our corporate IT department gave them all a deadline to update to the current operating system—they all complied or risked losing all connectivity to the network.
Almost every instrument vendor has told me that they can communicate with my laboratory system. I guess that is true: one talks in Russian and the other Sanskrit—they do communicate but is it effective? Talking is not necessarily communication!
I remember one open EIA machine that had a TCP/IP port but it was not functional by the standard protocols. One had to emulate a serial port to get some rudimentary communication. The port’s light blinked, however. I never imagined that someone would put a nonfunctional port as a mere decoration.
On the other hand, I have had excellent experience with another software vendor Medinfo. Even if the vendor did not have the interface developed, they could build it from scratch in a few weeks. Paradoxically, it was faster for these new interfaces than some so-called already interfaces.
I must emphasize: This is a collaborative team effort between the blood bank information system, software vendor, instrument vendor, and your institution’s IT staff. There must be excellent cooperation between them for a successful result.
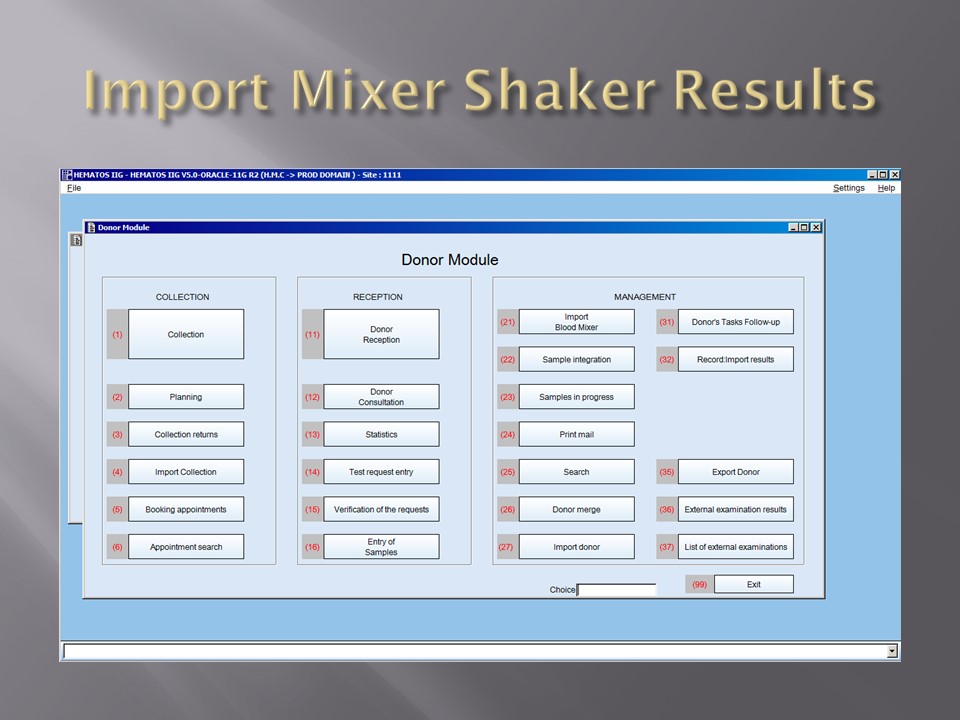
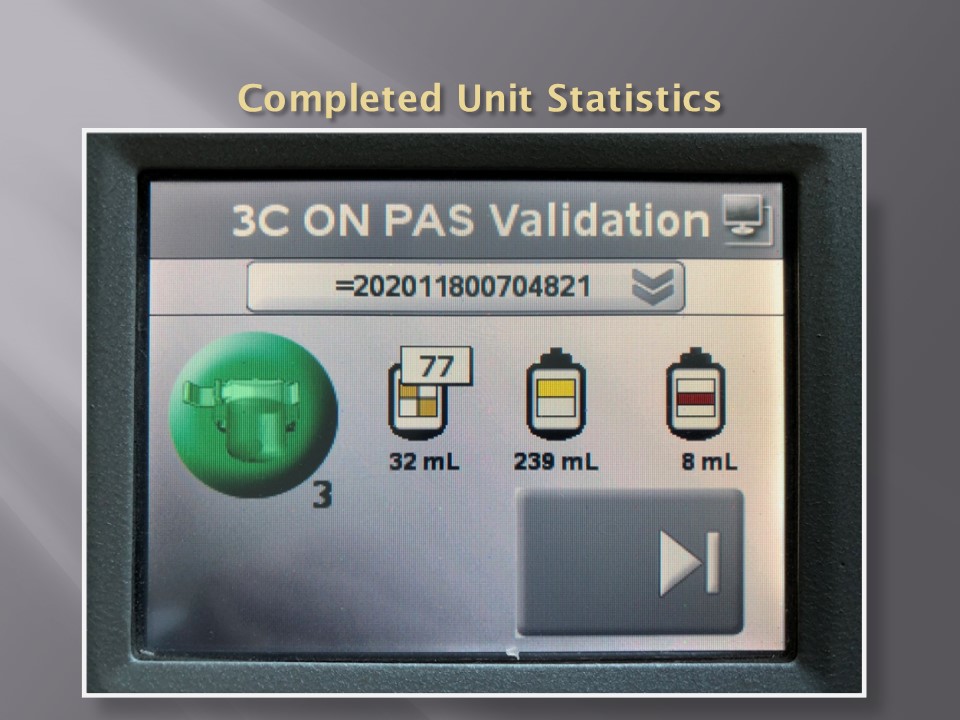
When installing the Medinfo Hematos IIG software, many of our most important interfaces (the Terumo mixed shaker, Trima, Reveos and its predecessor Atreus, Mirasol illuminator) had no developed interfaces when we started, This was a risk; but actually those interfaces were developed in a few weeks and fully functional. In fact, we were the first site in the world to have those interfaces working—and without any Middleware.
In general, the blood bank software vendor installed the completed interface and did some low-level testing. Then, my blood bank computer team did the testing. The final responsibility for testing and acceptance was with the end-user blood bank team.
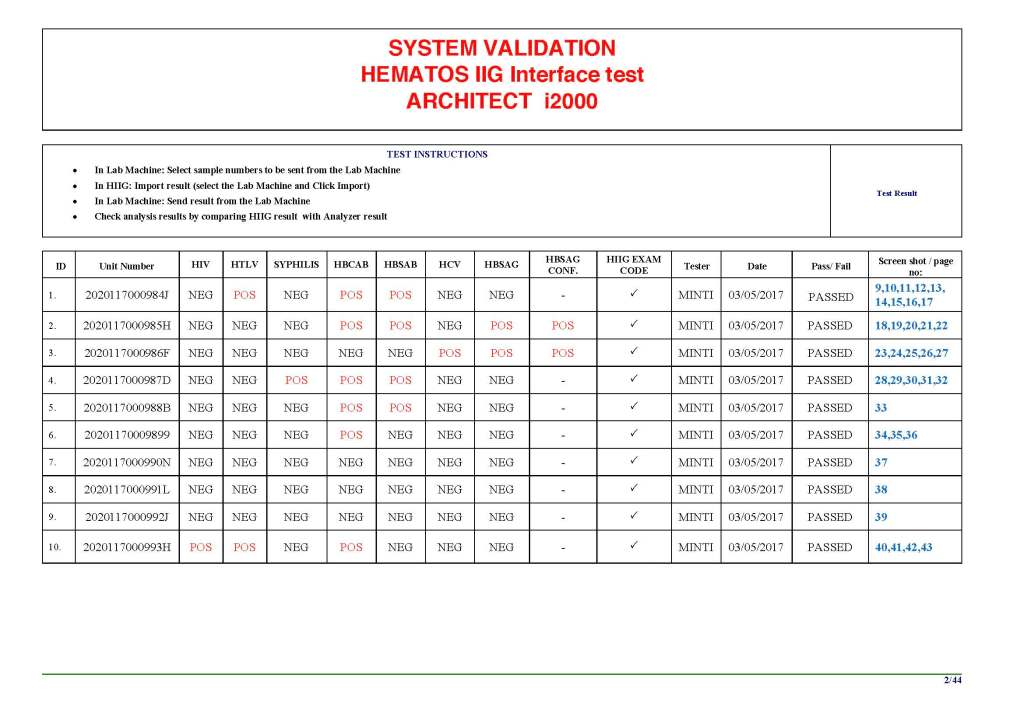
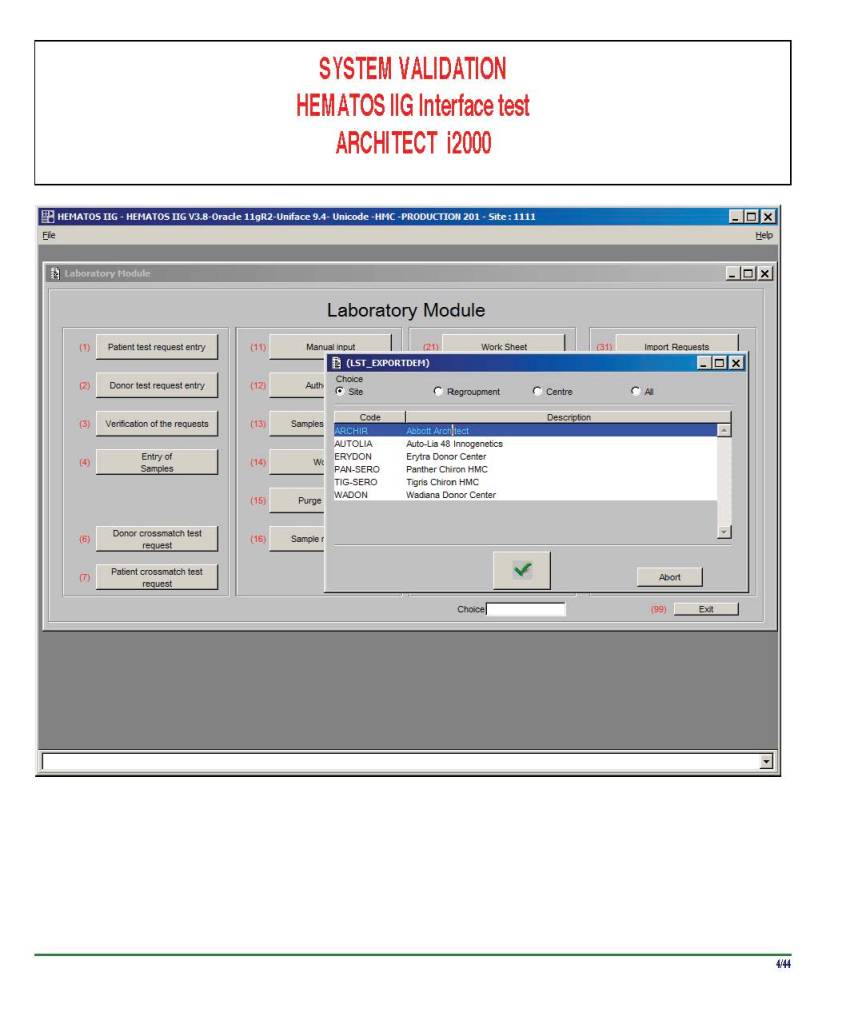
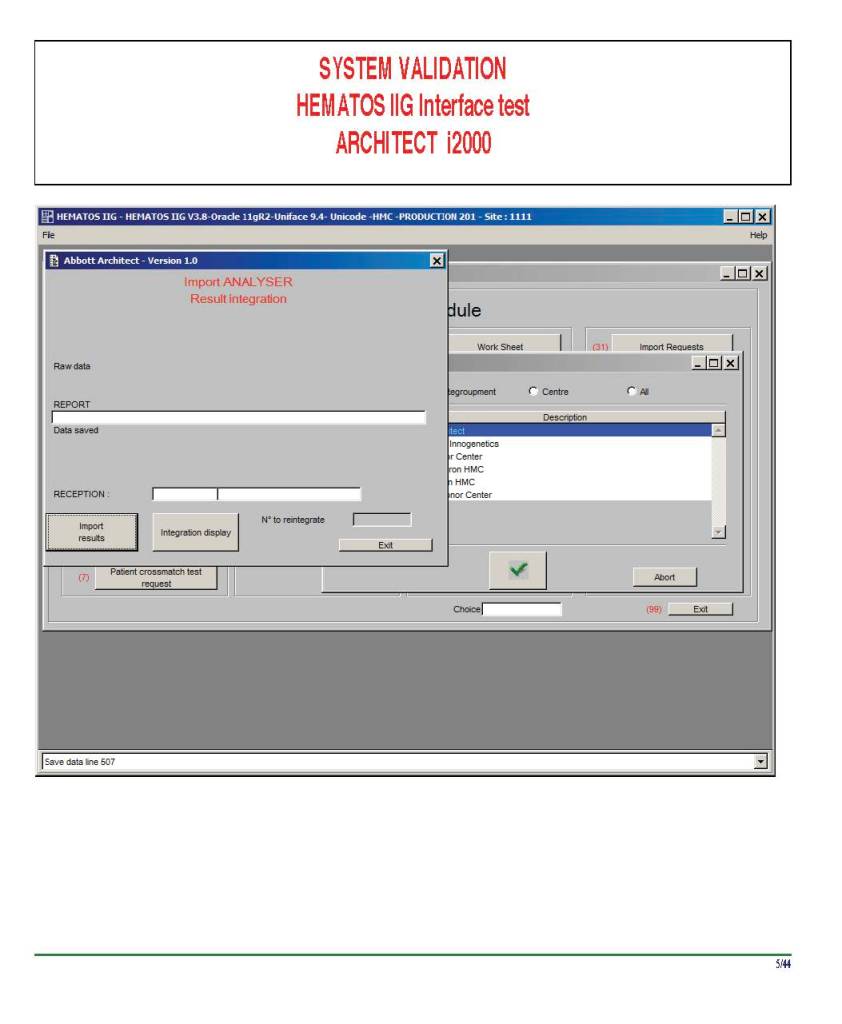
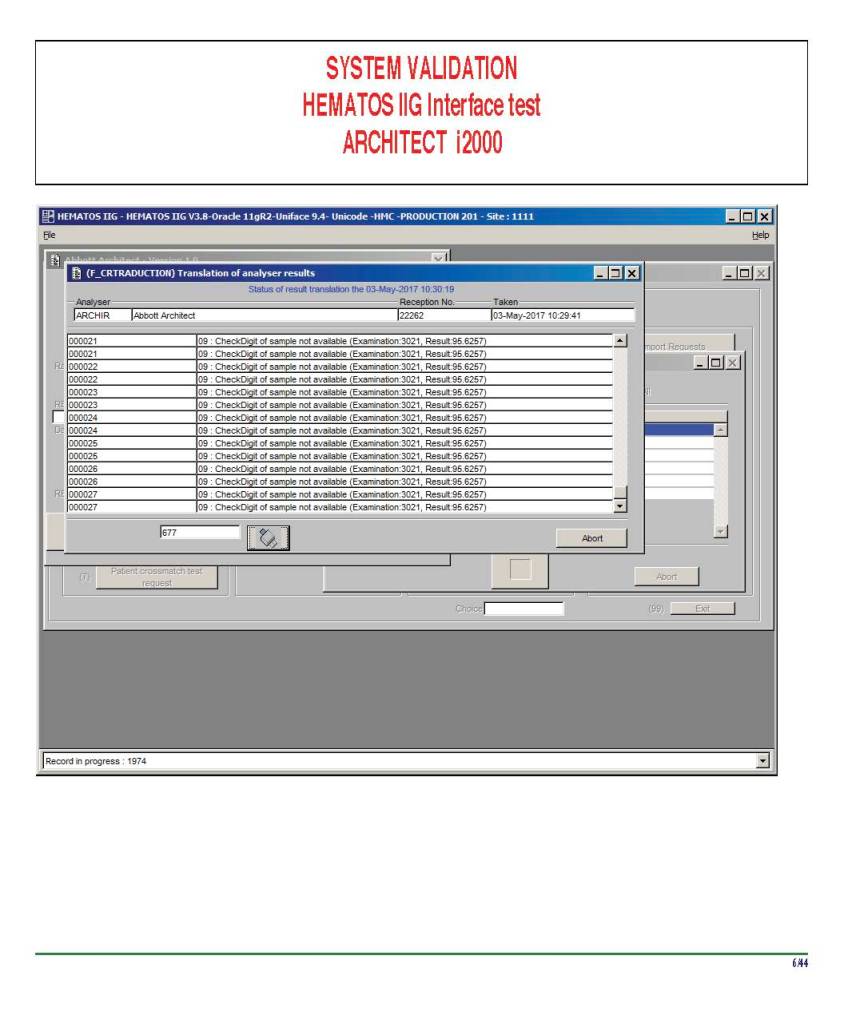
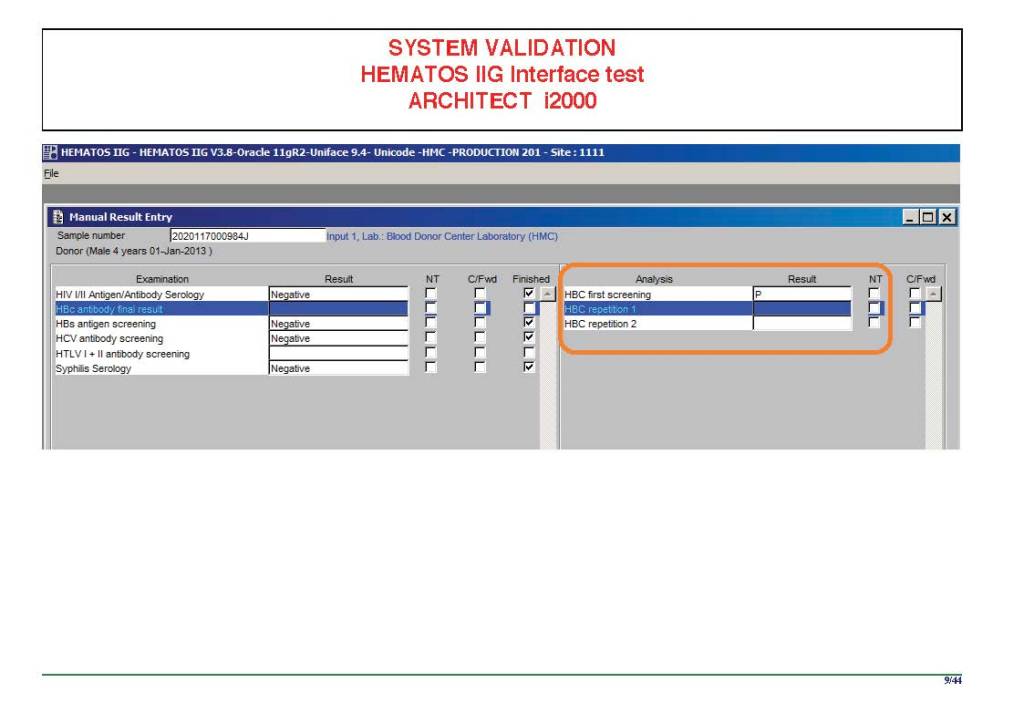
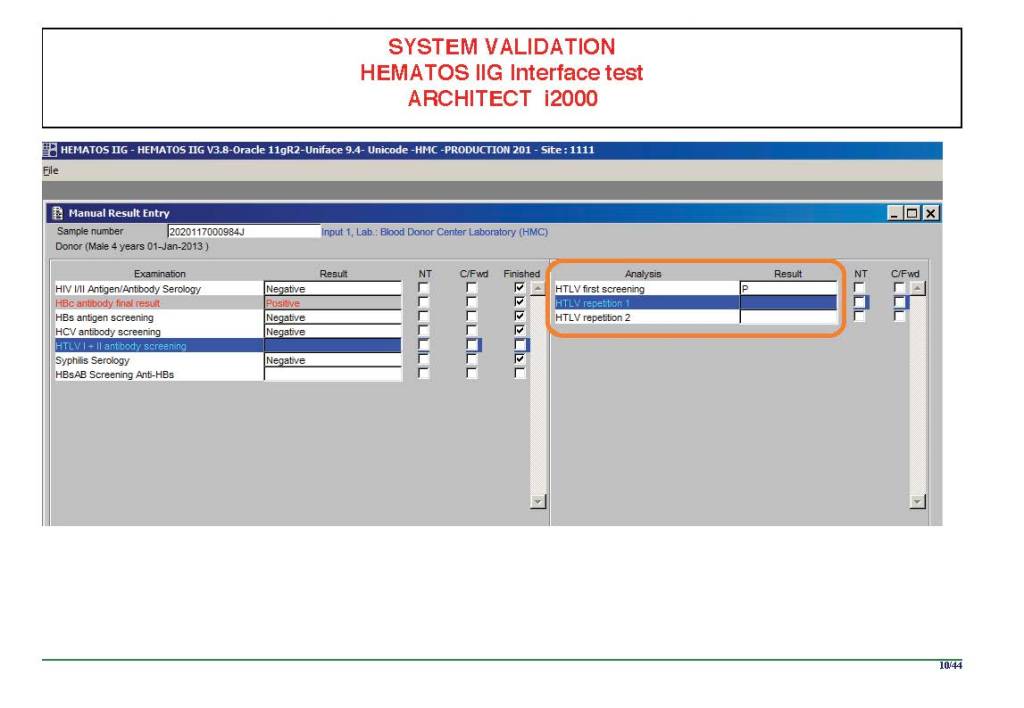
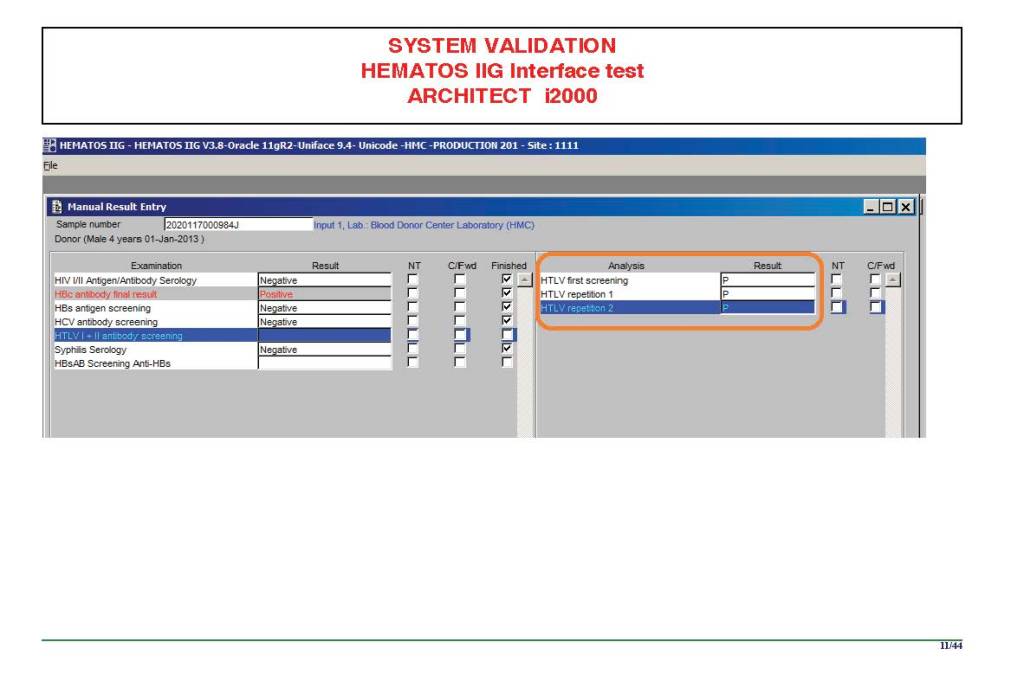
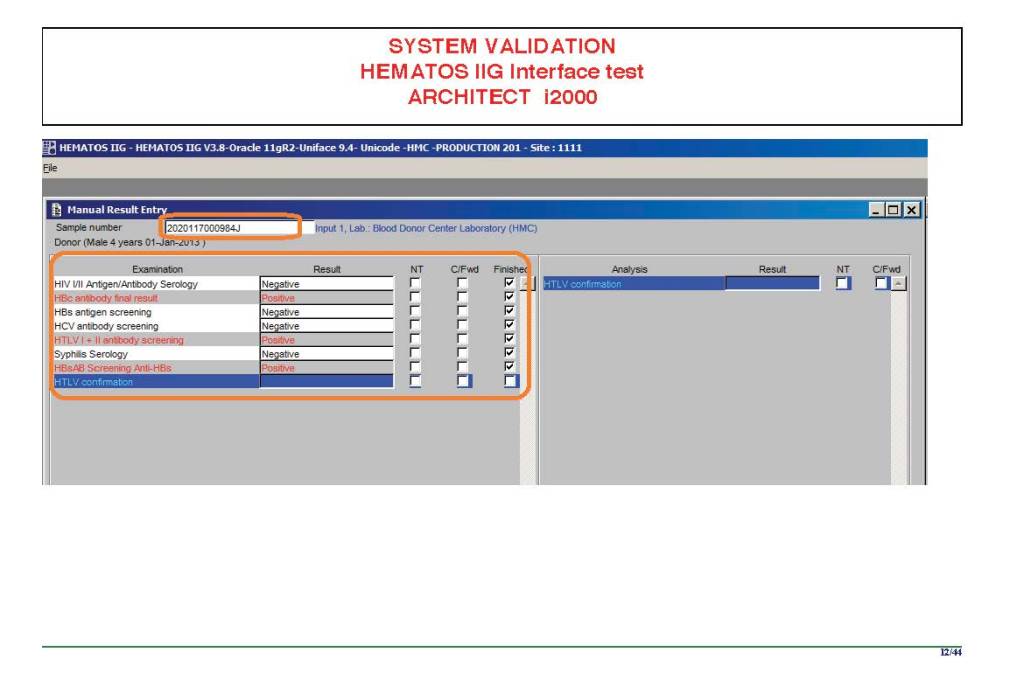
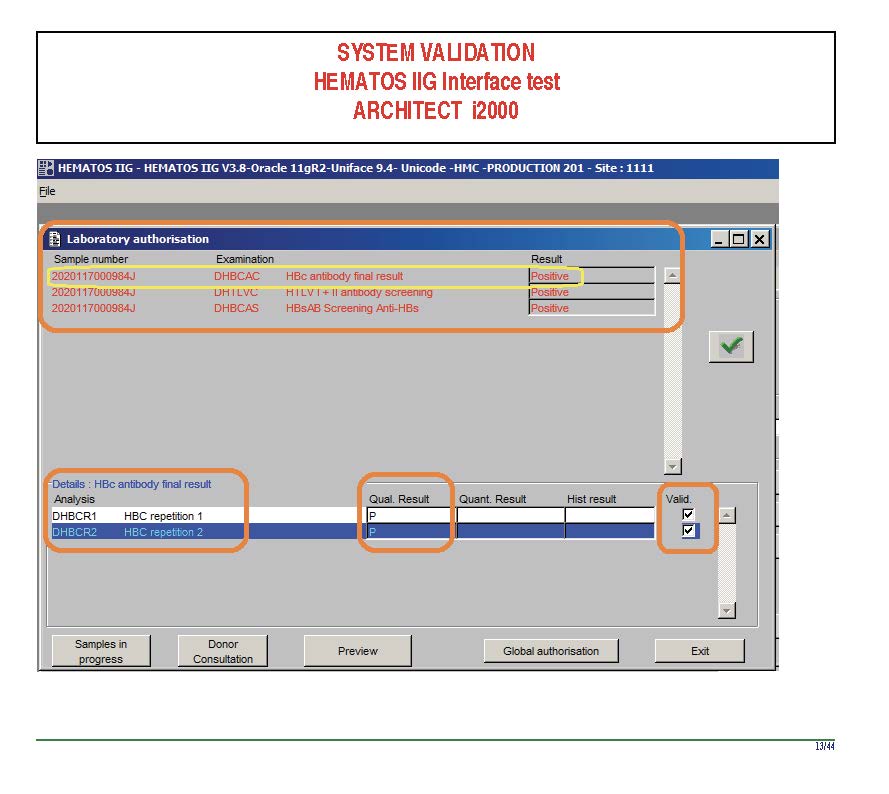
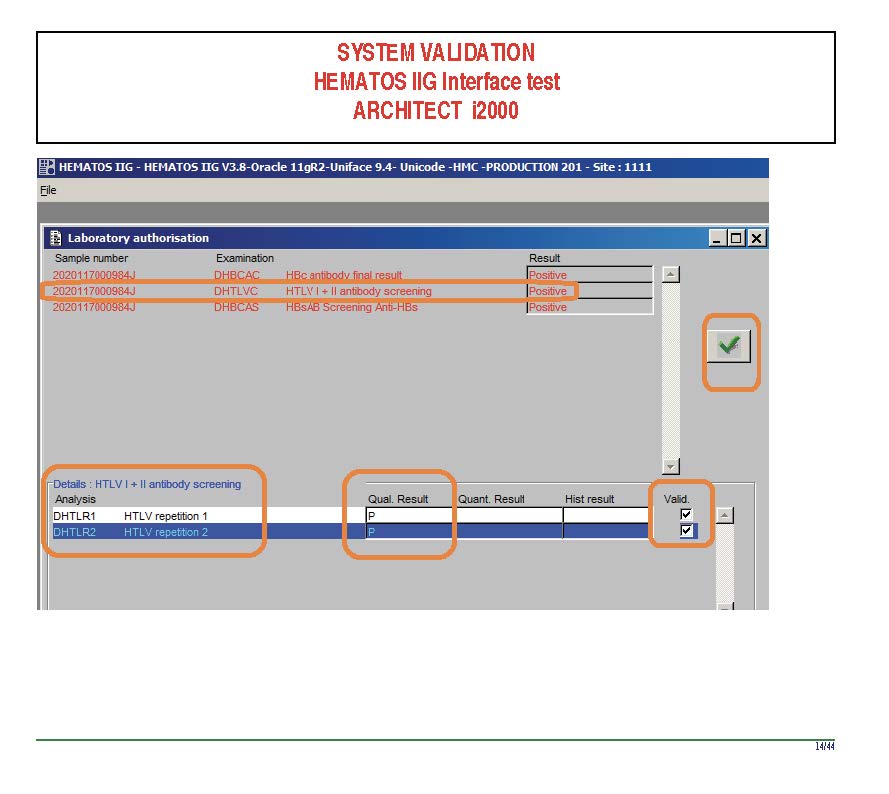
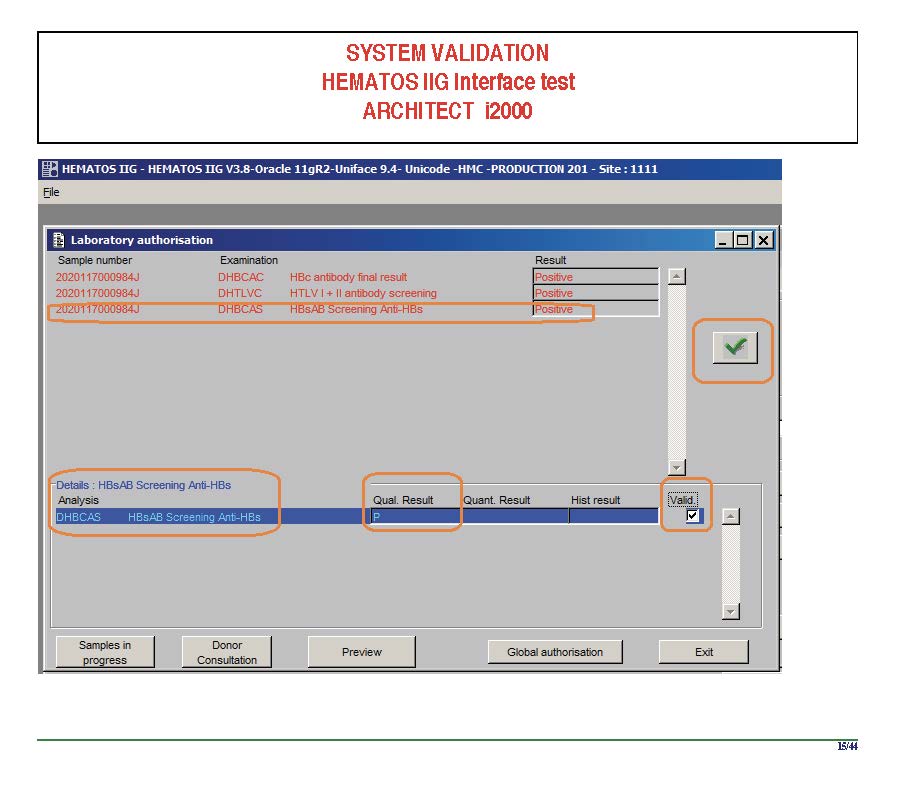
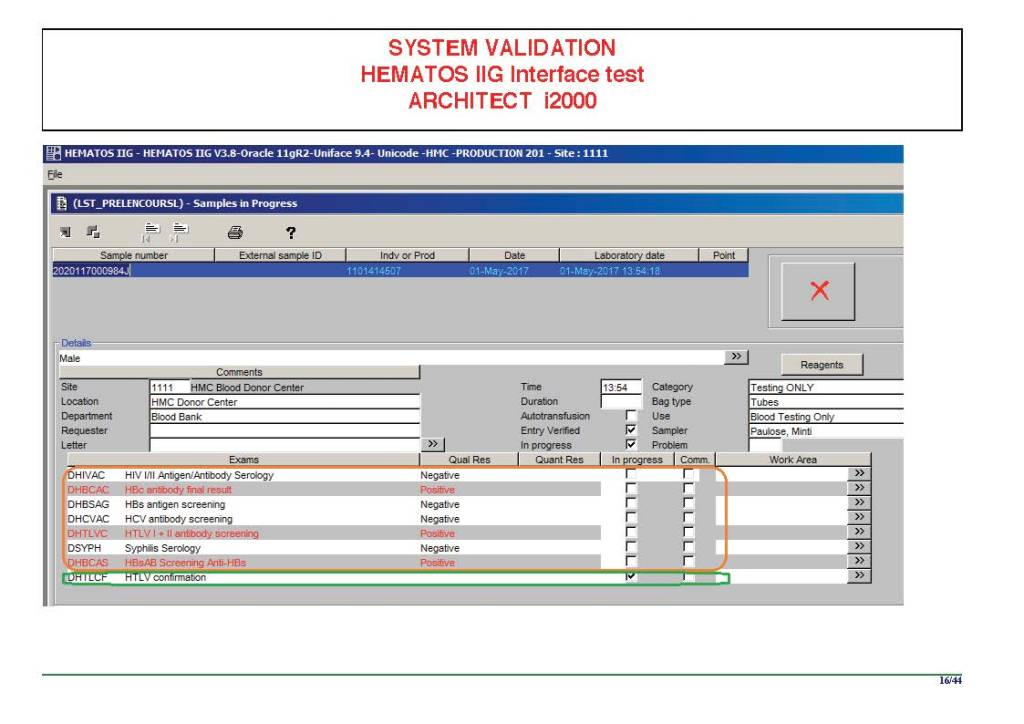
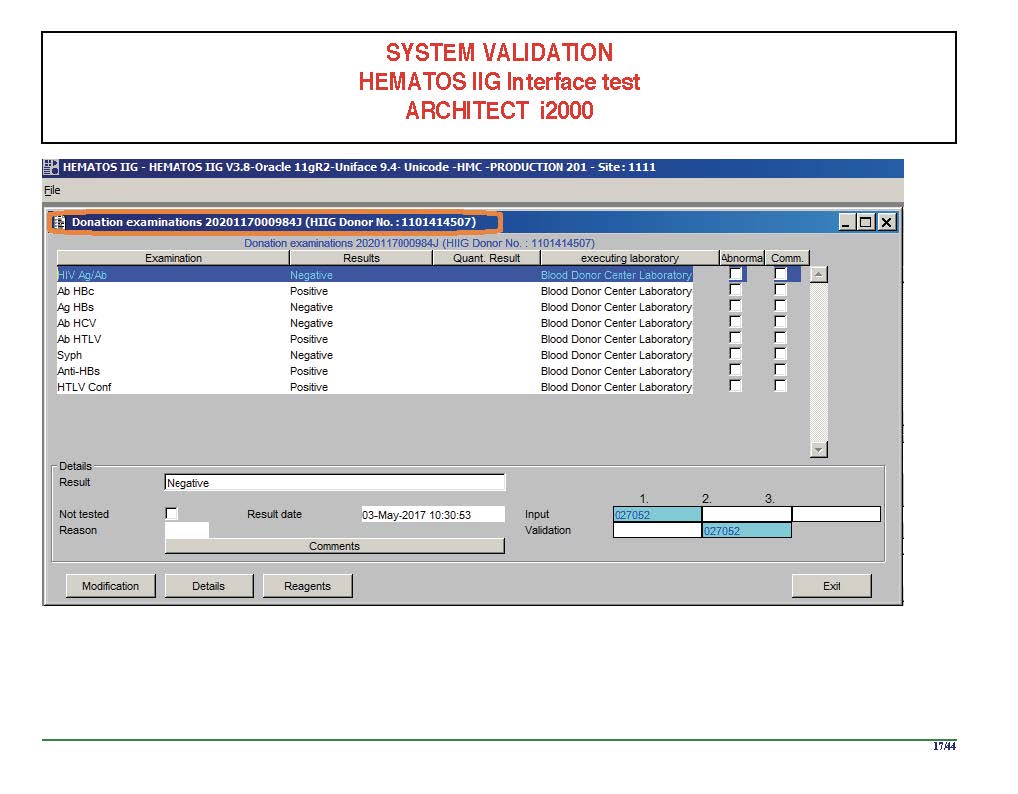
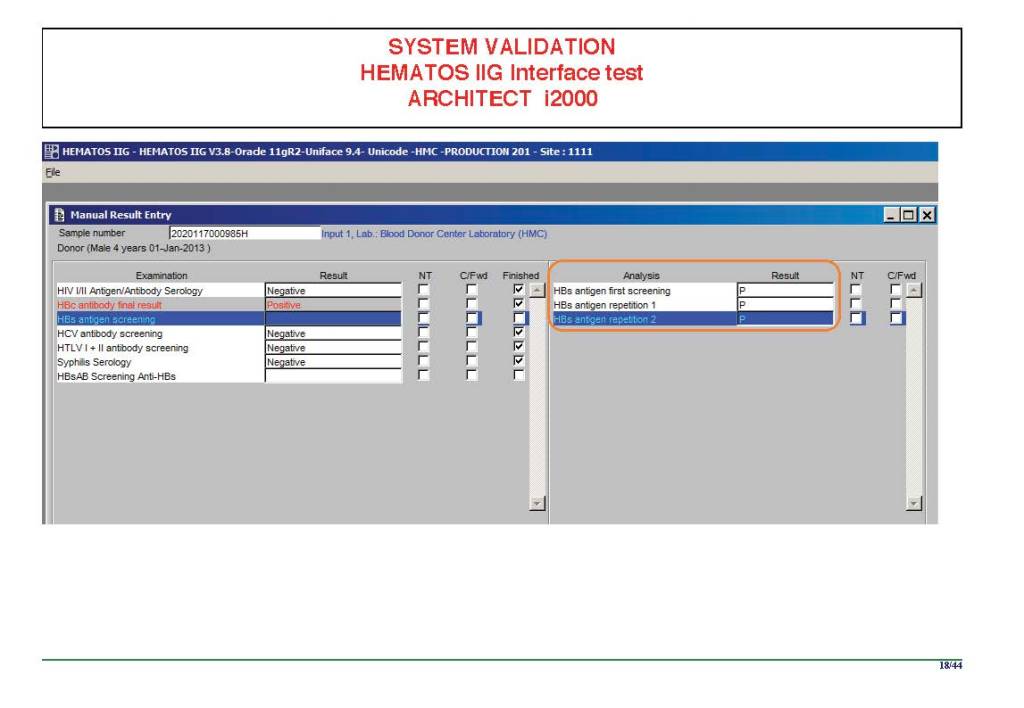
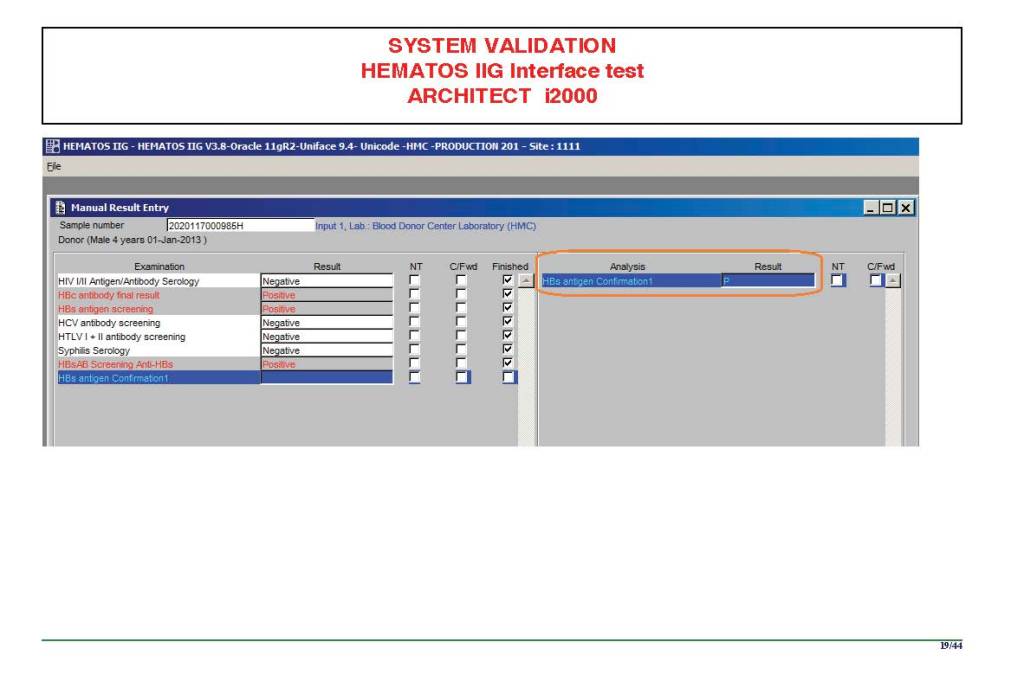
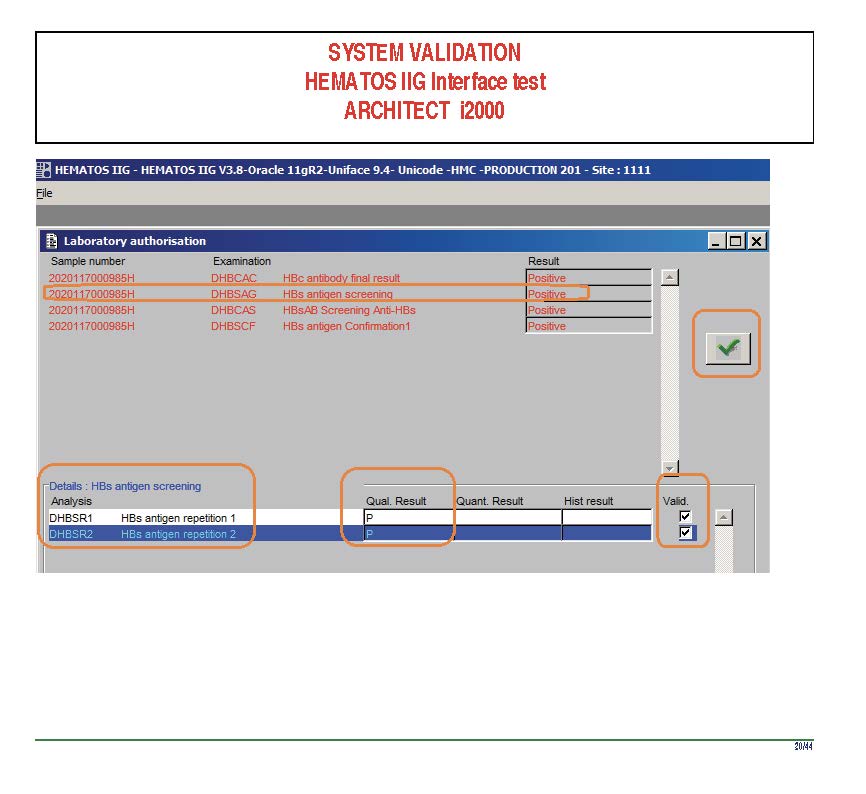
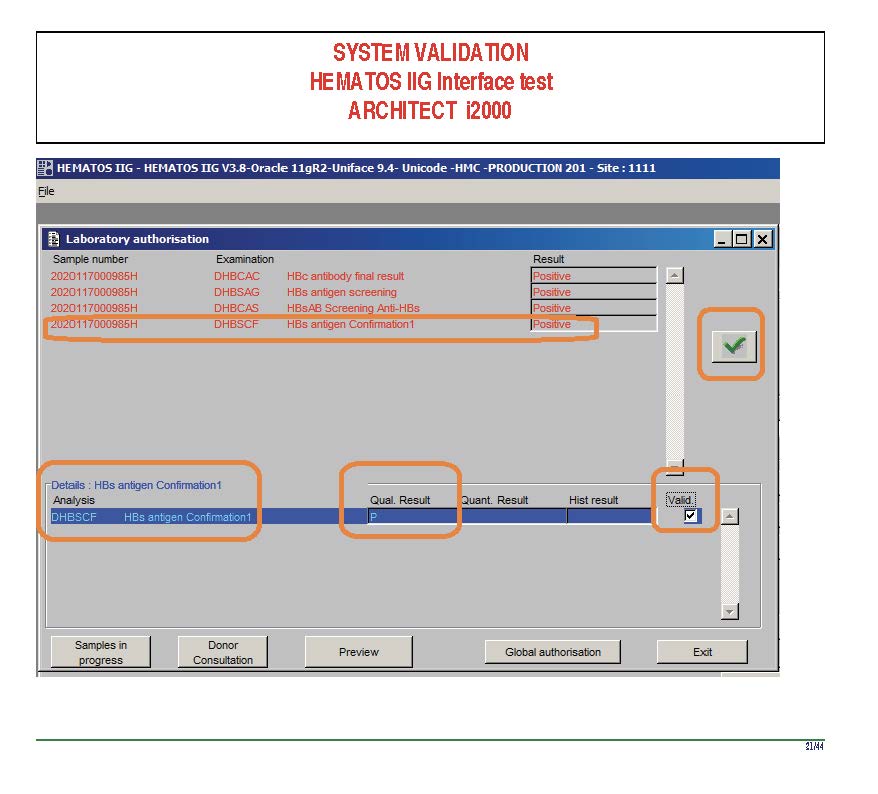
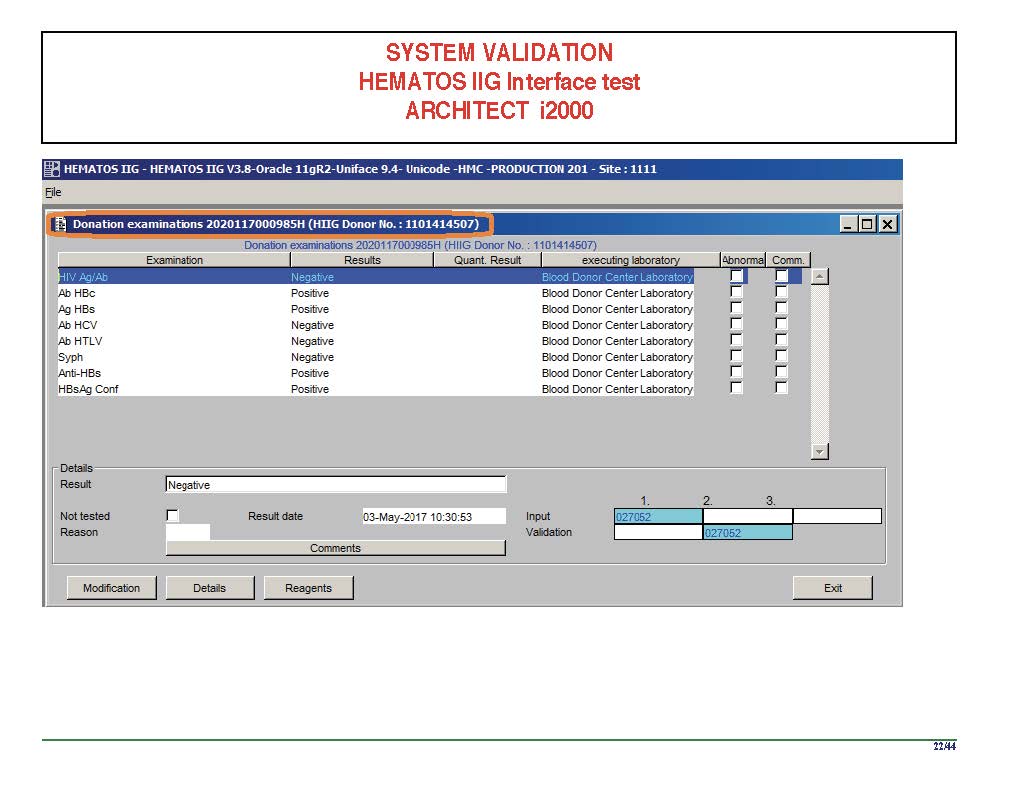
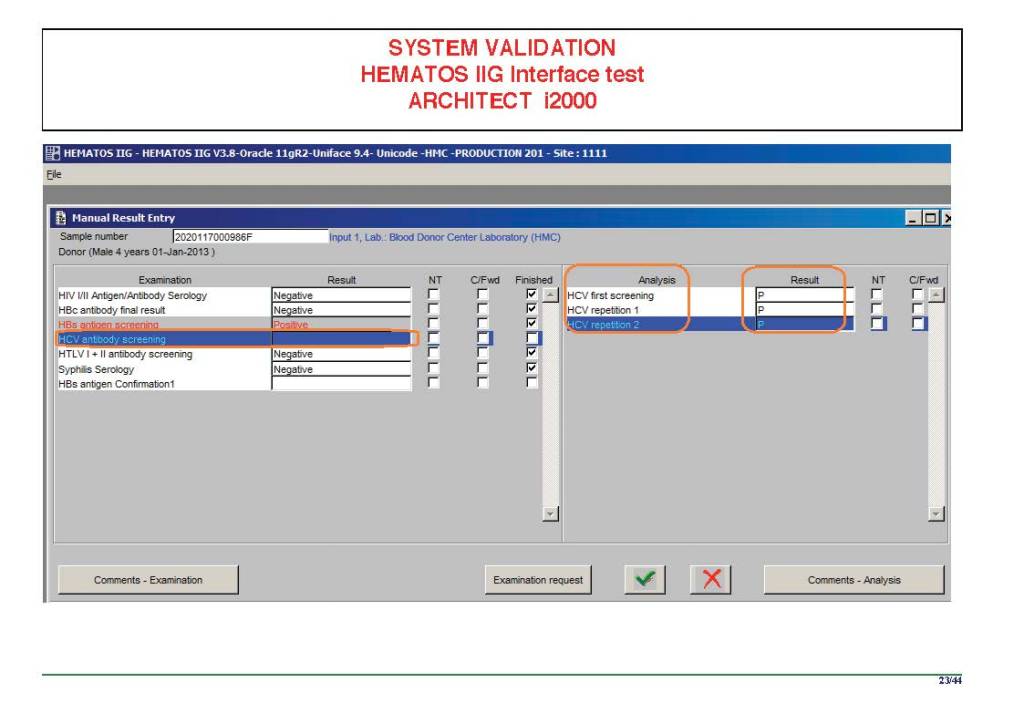
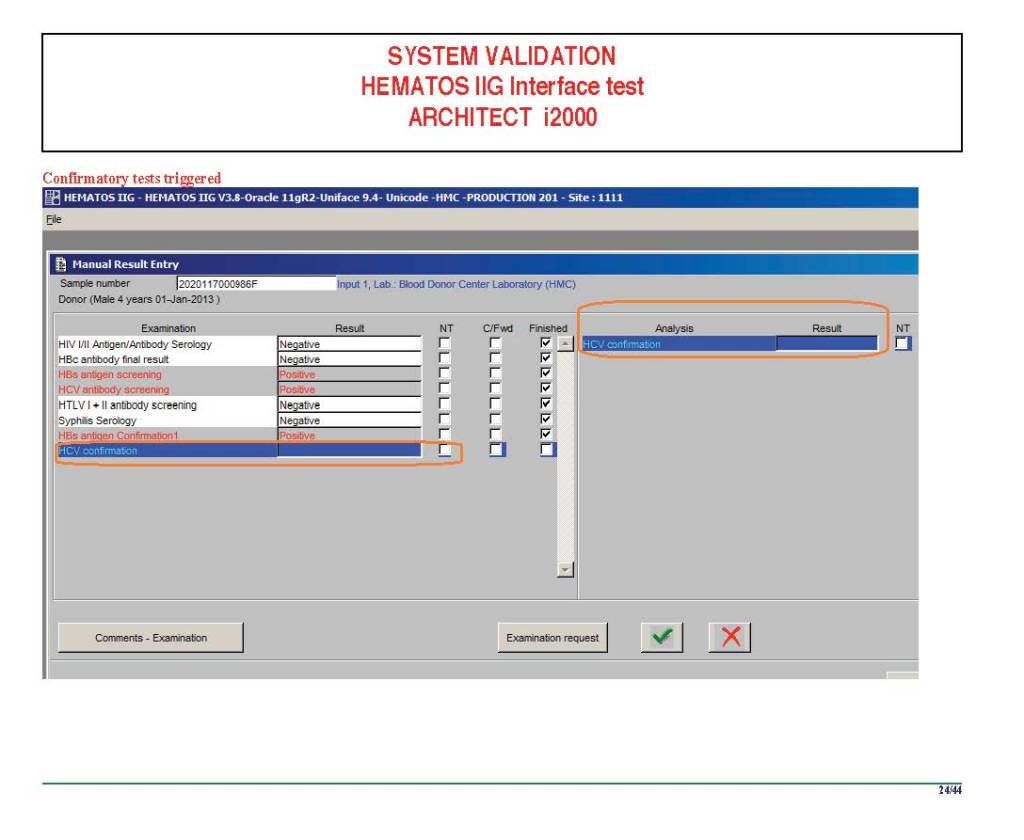
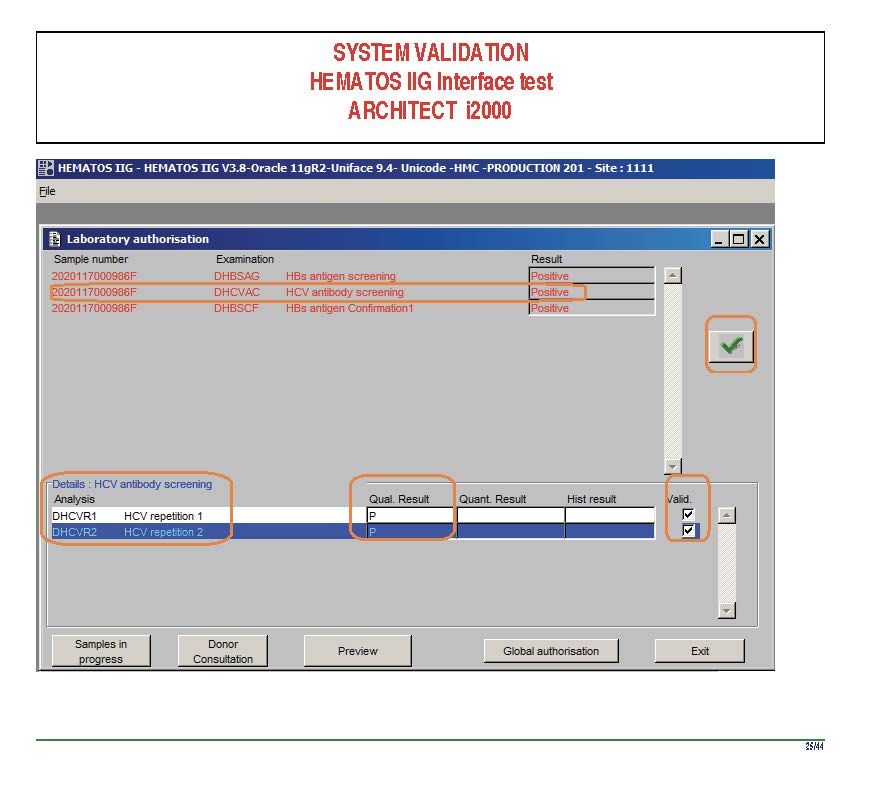
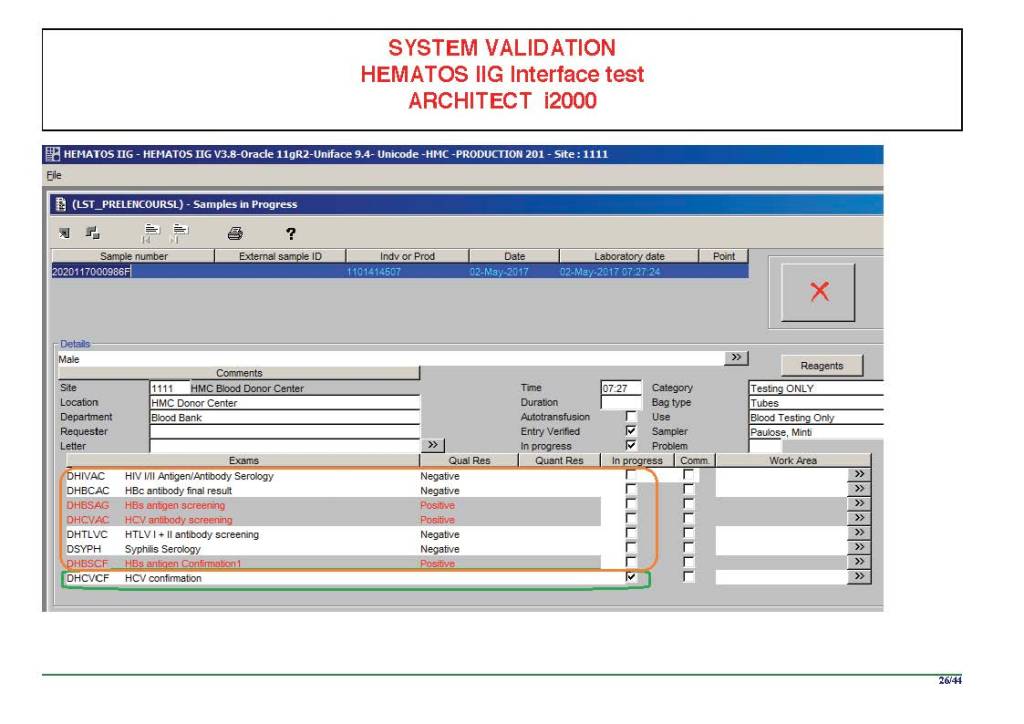
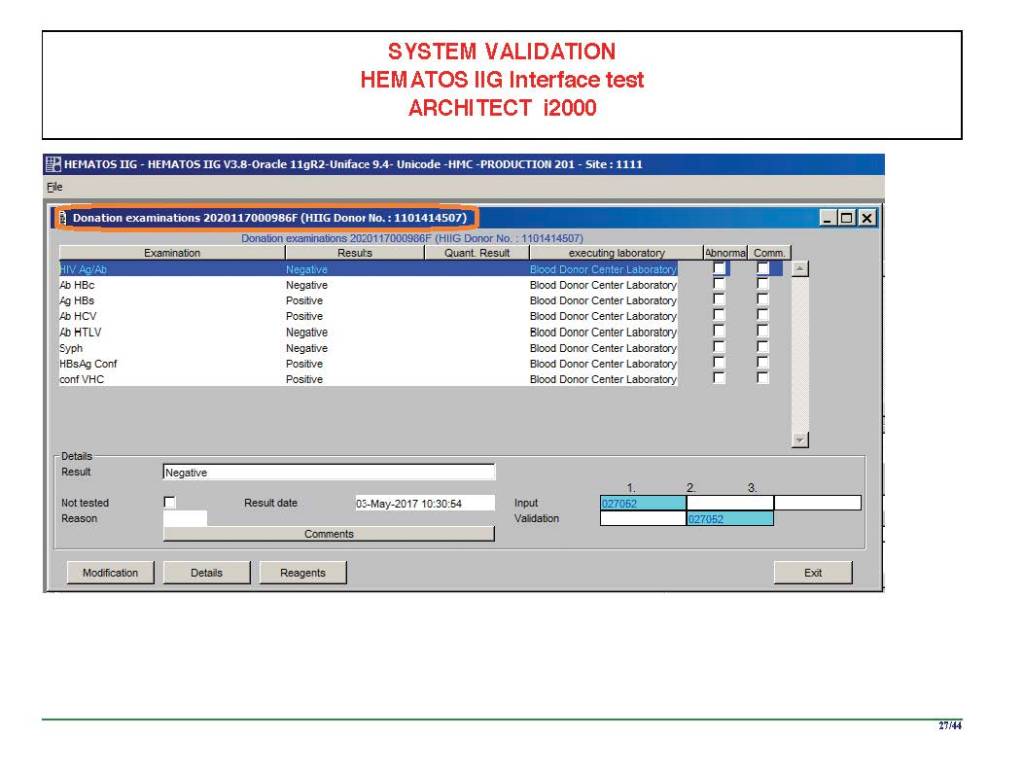
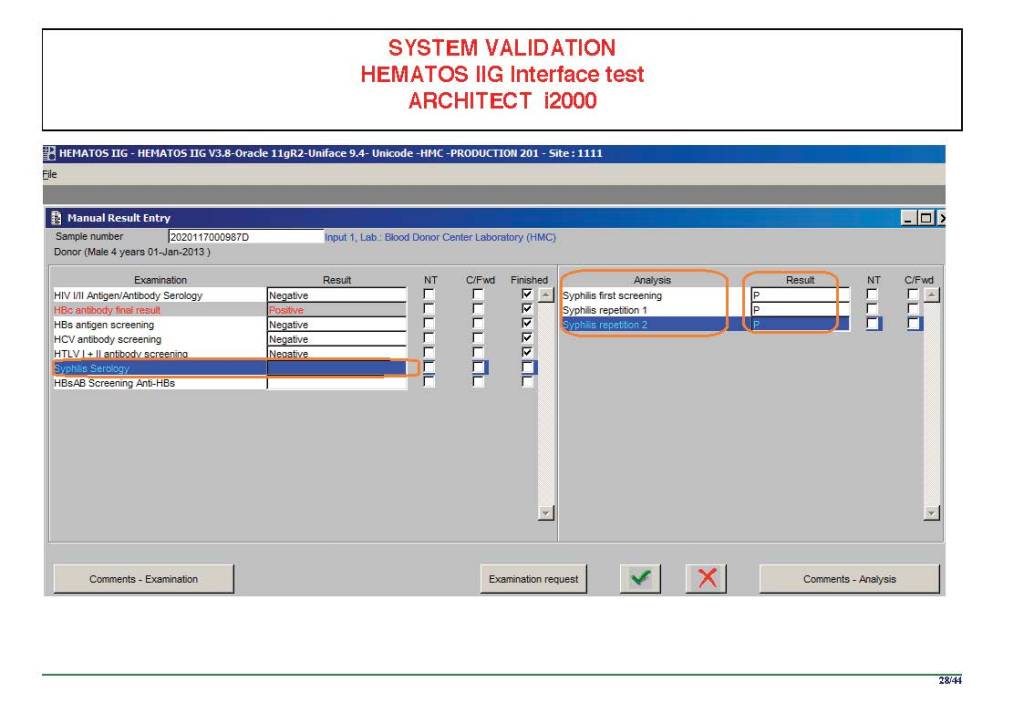
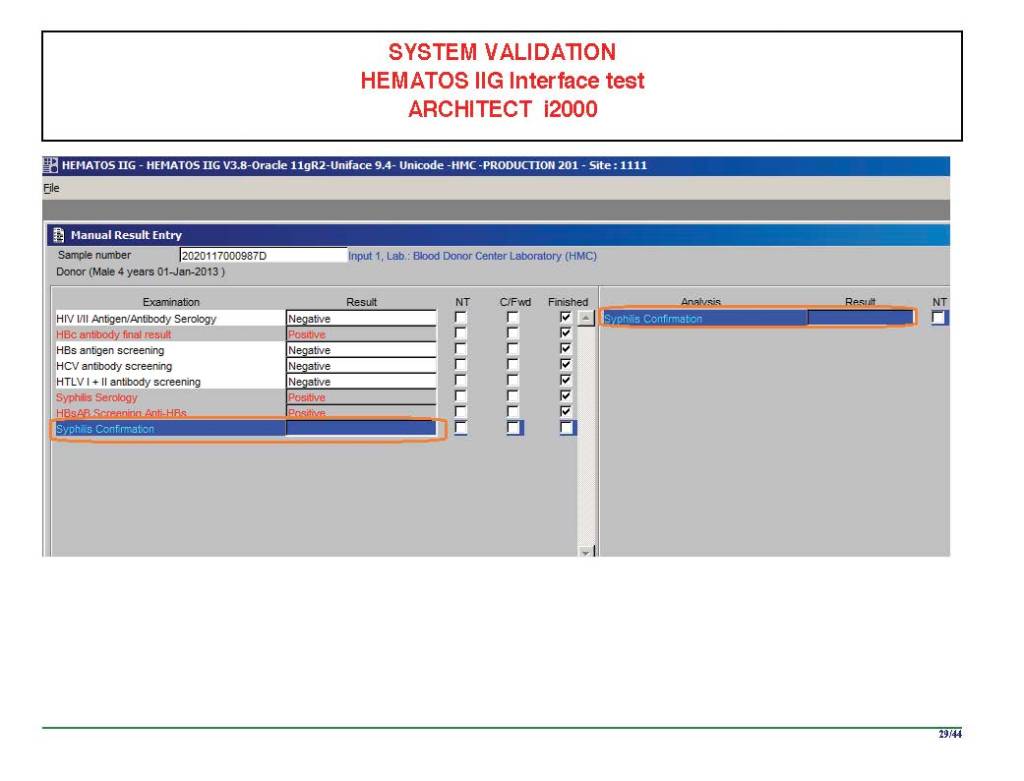
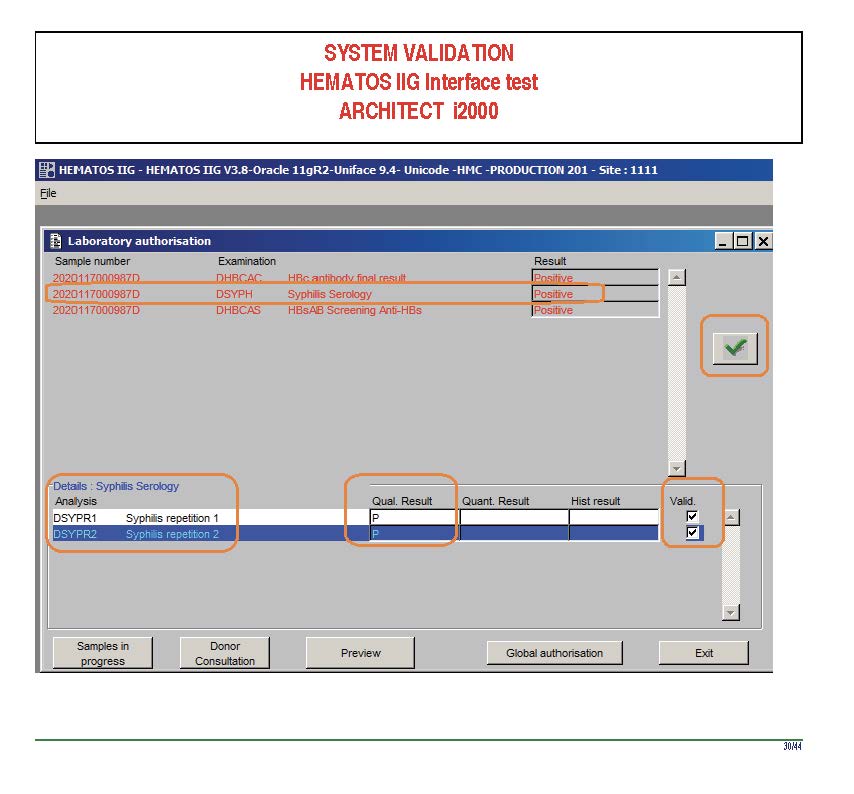
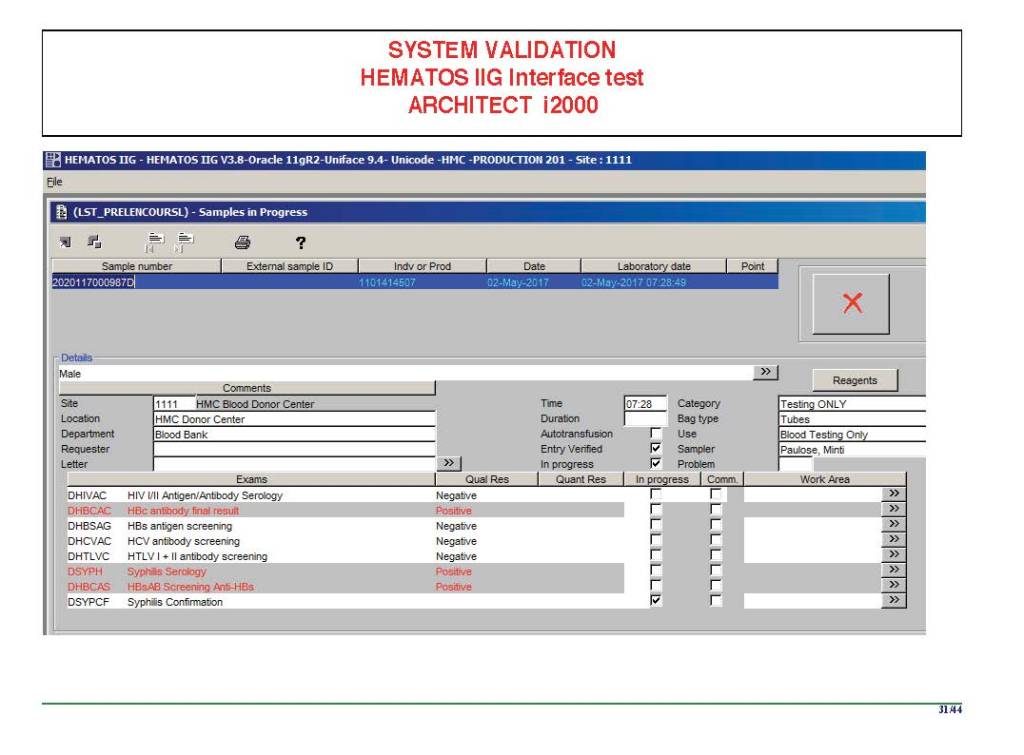
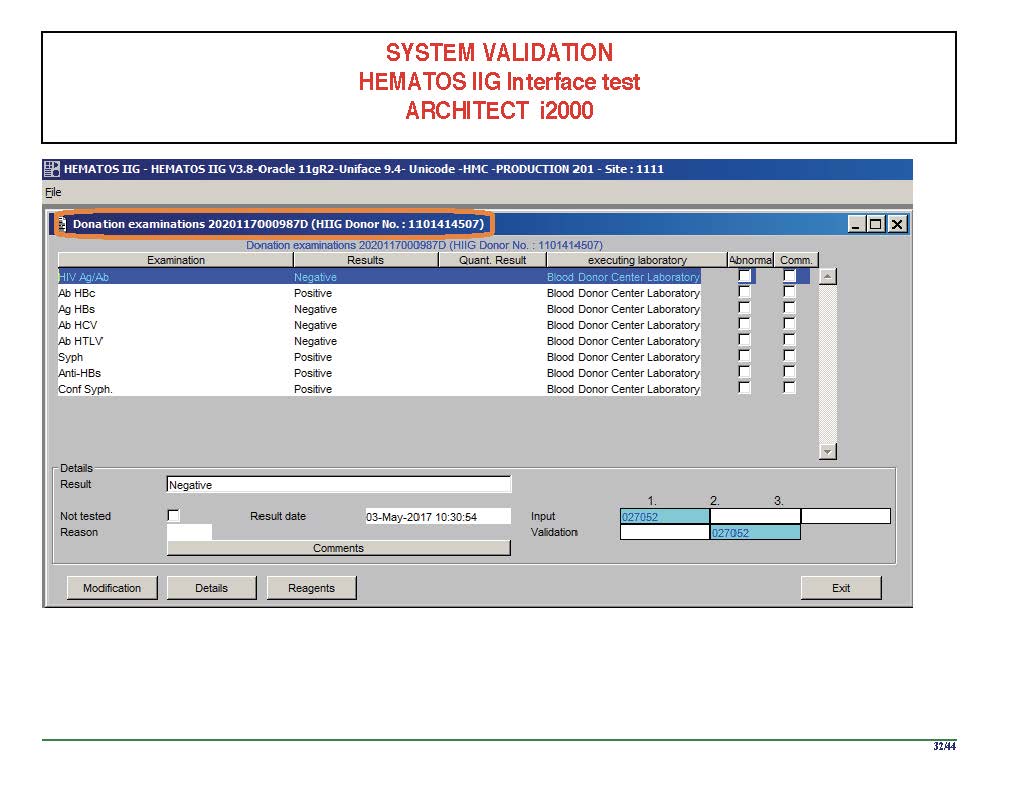
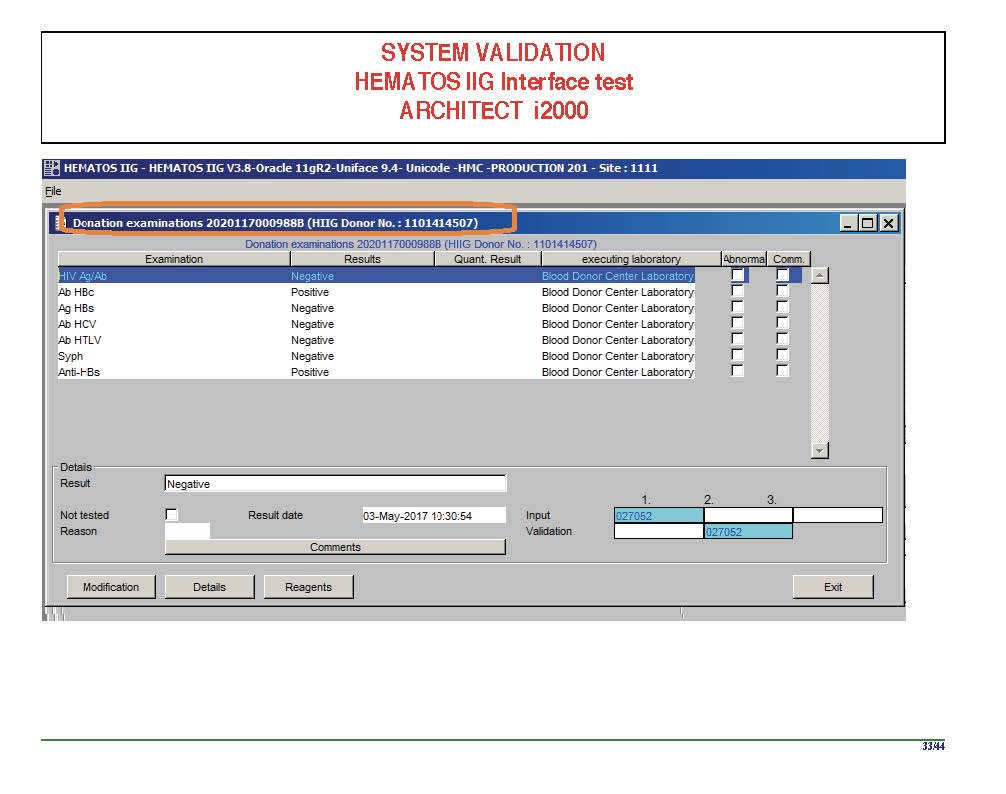
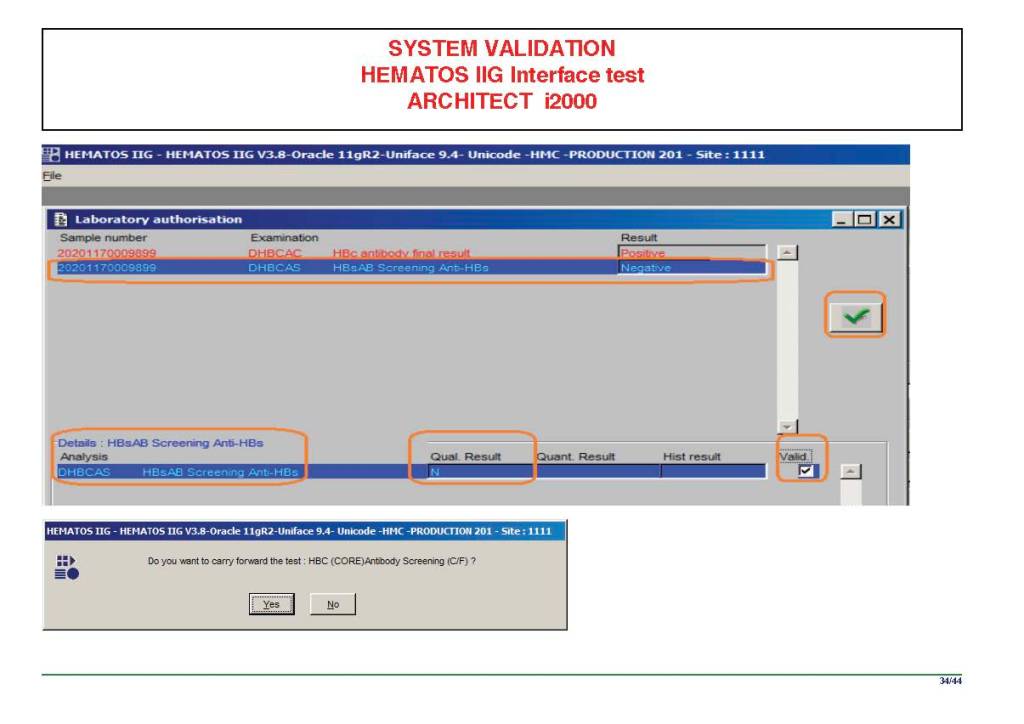
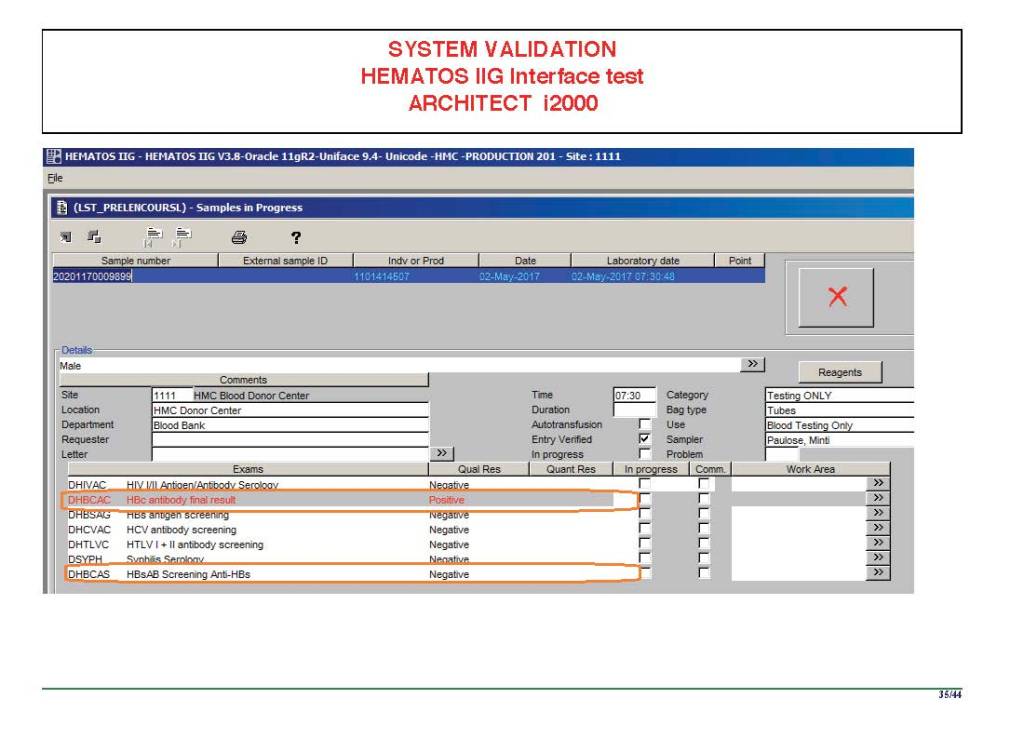
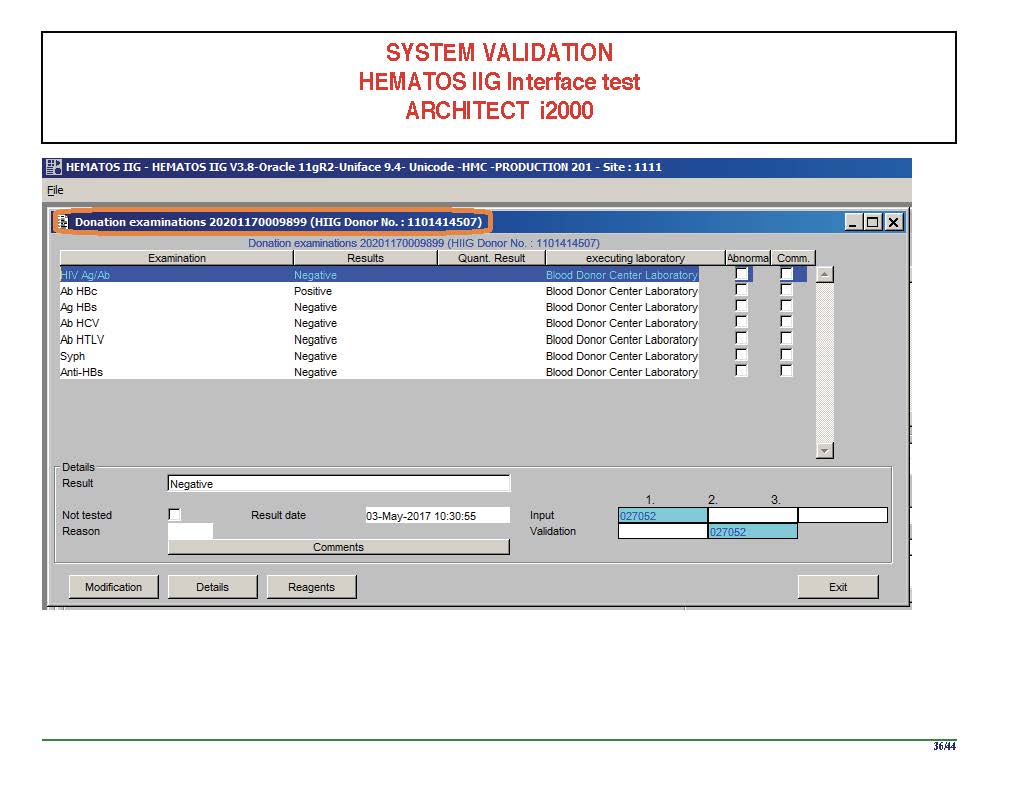
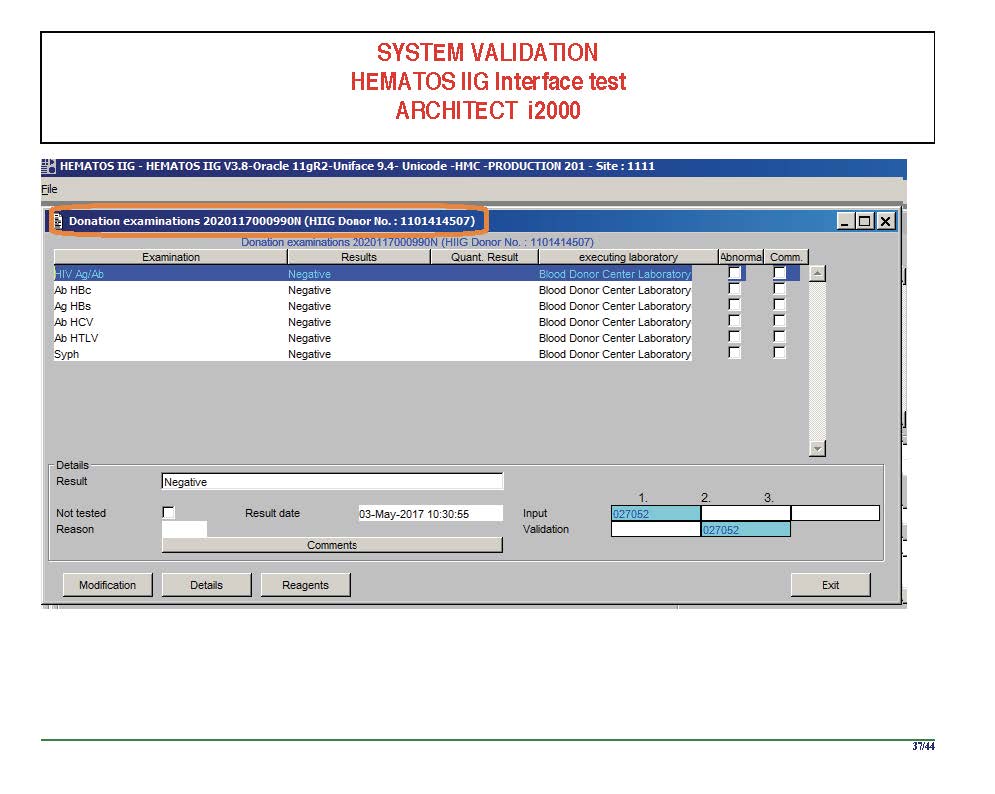
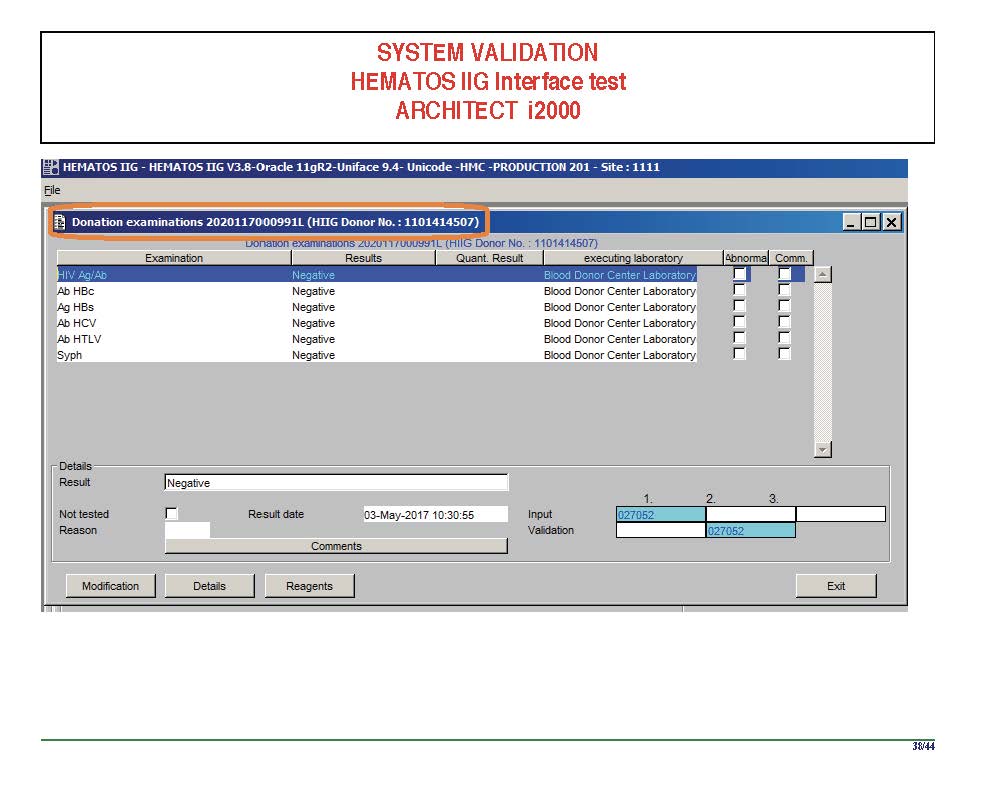
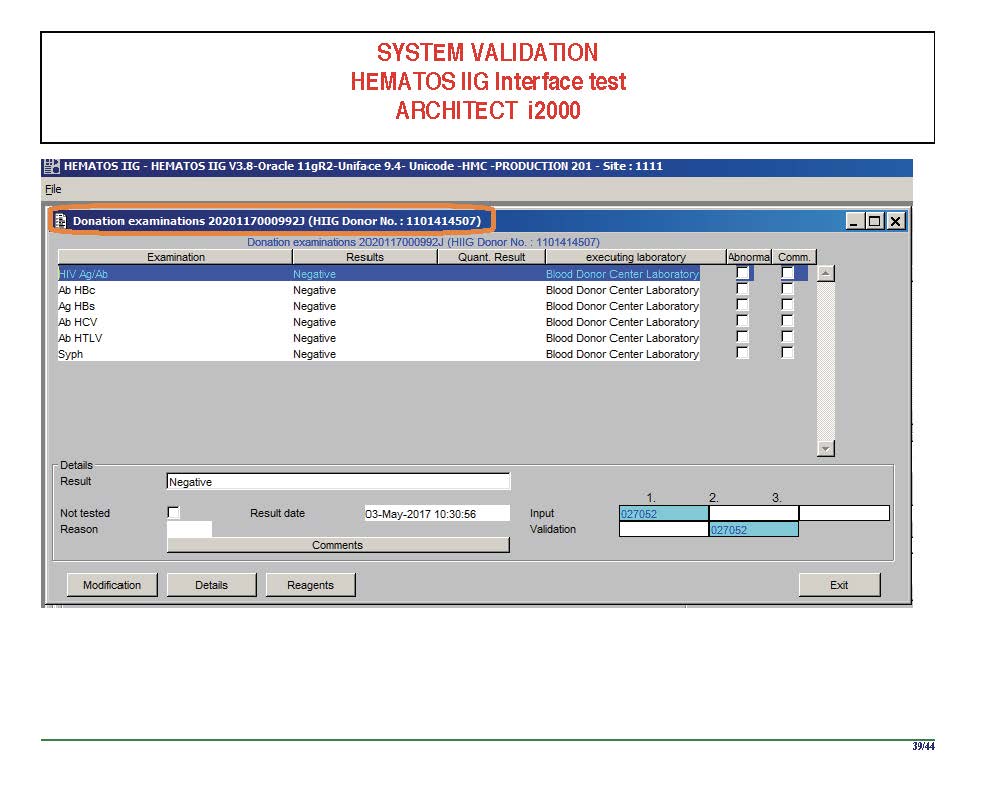
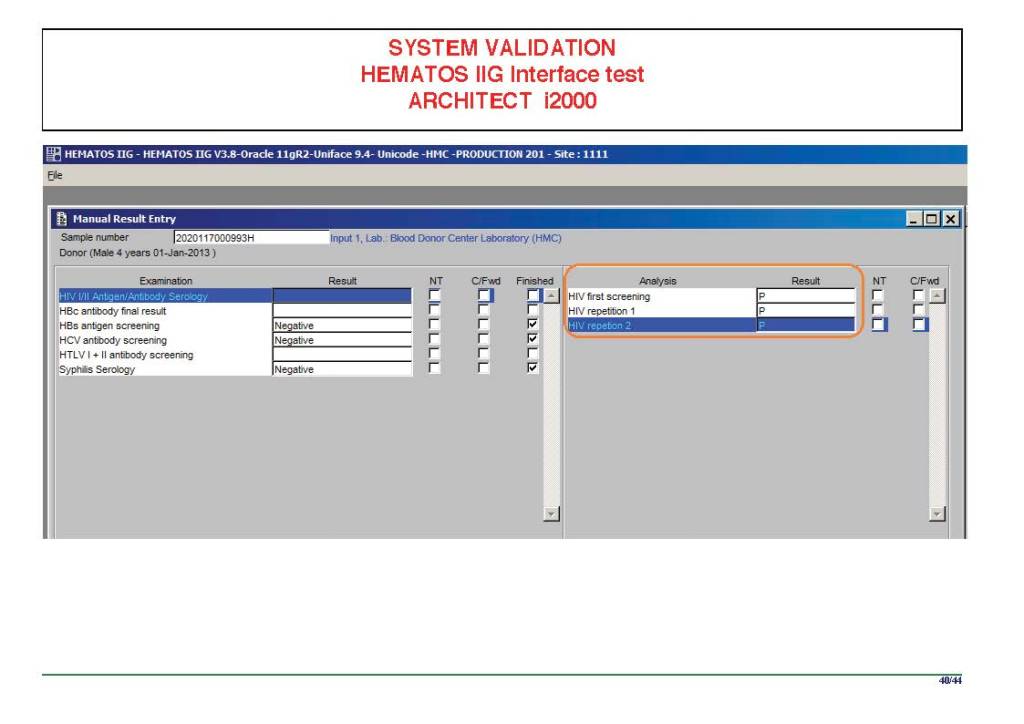
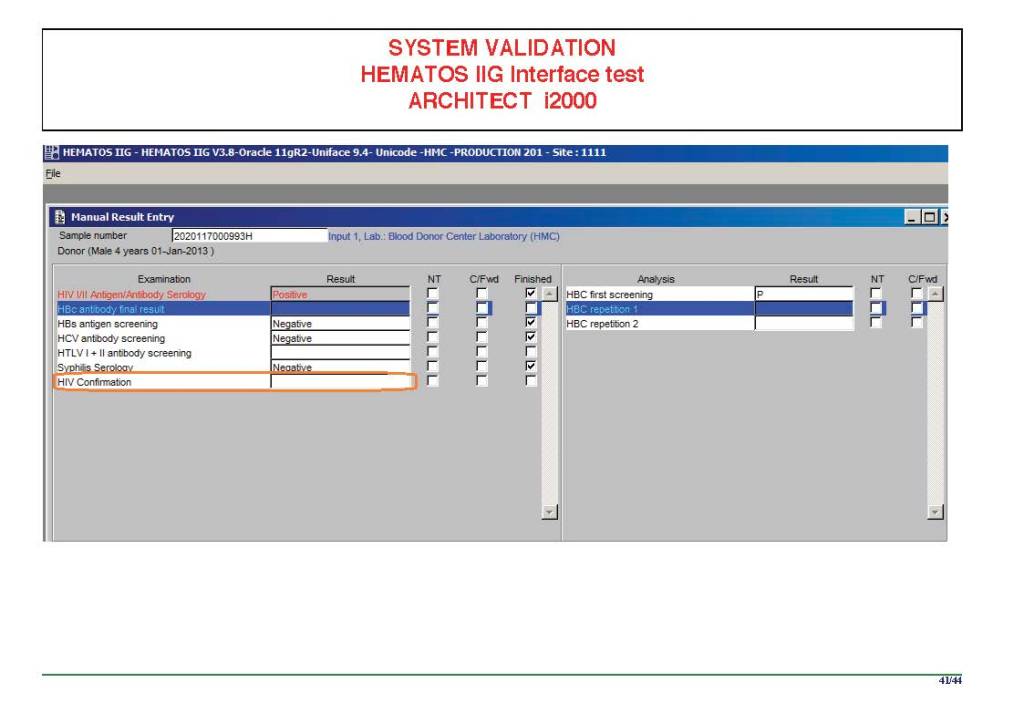
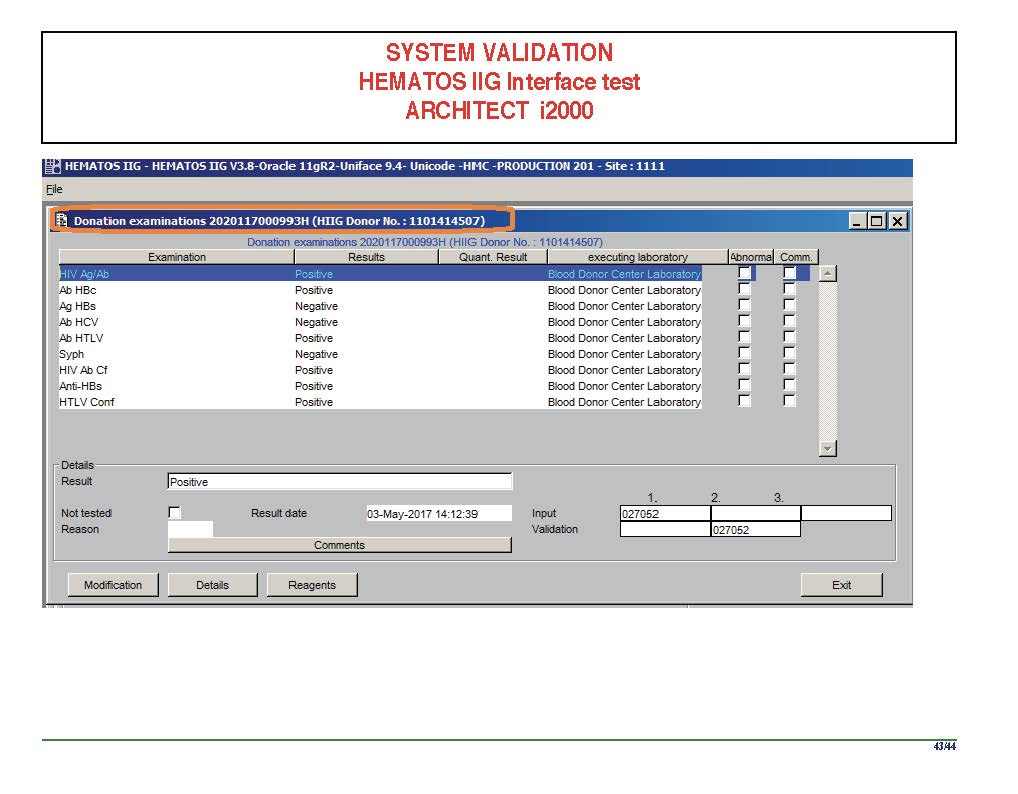
I am attaching a copy of our Abbott Architect interface as updated a few years ago. Again, here I wrote the validation protocol and assigned the tasks to the Medinfo Super Users. To perform this EIA testing, we still had to register donors and collect and then export the specimens to the donor marker testing laboratory before the actual interface testing could begin. This was all done in a special test domain separate from the production domain.
I made the validation criteria and reviewed all data as Division Head of Laboratory Information Systems. Representative screen shots were made. All data was sent to me. My final acceptance was required before the interface could be activated.
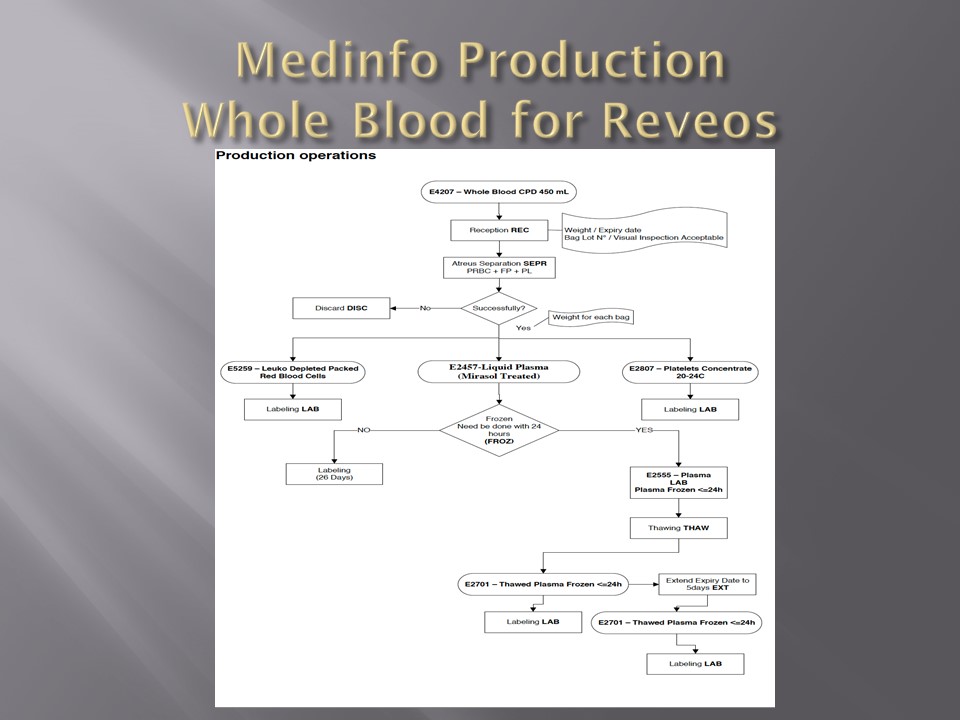
Automated component processing (Reveos) and component modification are more complicated and will be covered in a future post.